

MXene作为一类二维过渡金属碳化物/氮化物材料,凭借其高导电性、可调表面官能团和丰富的活性位点,在电催化领域展现出巨大潜力。
以下结合密度泛函理论(DFT)计算,详述其在氢析出反应(HER)、氮还原反应(NRR)、二氧化碳还原反应(CO2RR)等方向的具体应用案例及分析方法。
一、MXene在电催化中的DFT计算应用案例
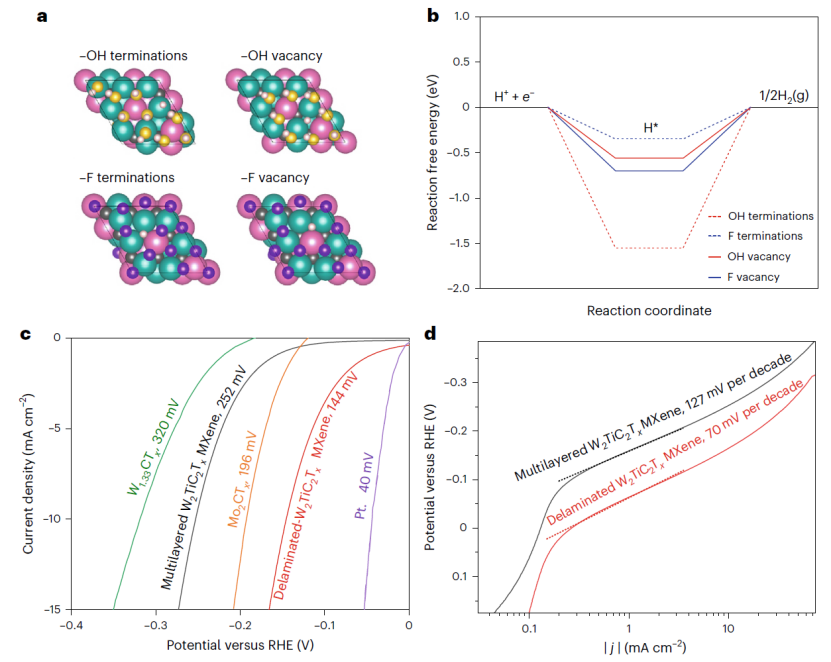
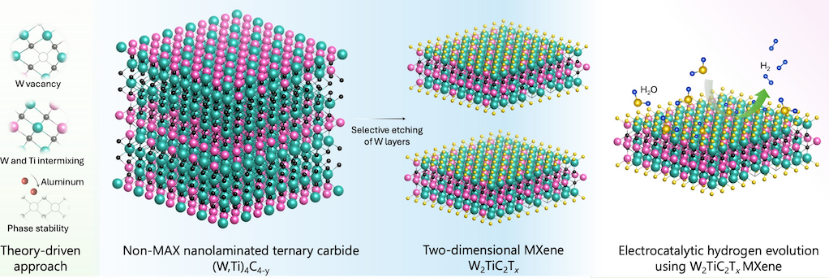
在氢析出反应(HER)相关研究中,针对传统 MXene 基于 MAX 相前驱体合成,而钨基 MAX 相因热力学不稳定性难以制备的问题,Anasori 团队通过理论指导,设计非 MAX 前驱体(W,Ti)₄C₄-y,成功合成 W₂TiC₂Tₓ MXene。在 DFT 分析中,活性位点识别显示,羟基(-OH)和氟(-F)终止的表面分别通过钨顶位(吸附能 – 1.55 eV)和三重位(-0.37 eV)吸附氢,且氧空位进一步优化吸附能为 – 0.77 eV;其电子结构方面,W₂TiC₂Tₓ的基面富集 W 原子,W 的 d 带中心位置接近费米能级,增强了 H * 吸附 / 脱附动力学;性能预测上,理论预测该材料 HER 过电位为 144 mV(10 mA/cm²),优于多数 MXene,且实验验证与计算结果一致。

DOI:https://doi.org/10.1038/s44160-025-00773-z
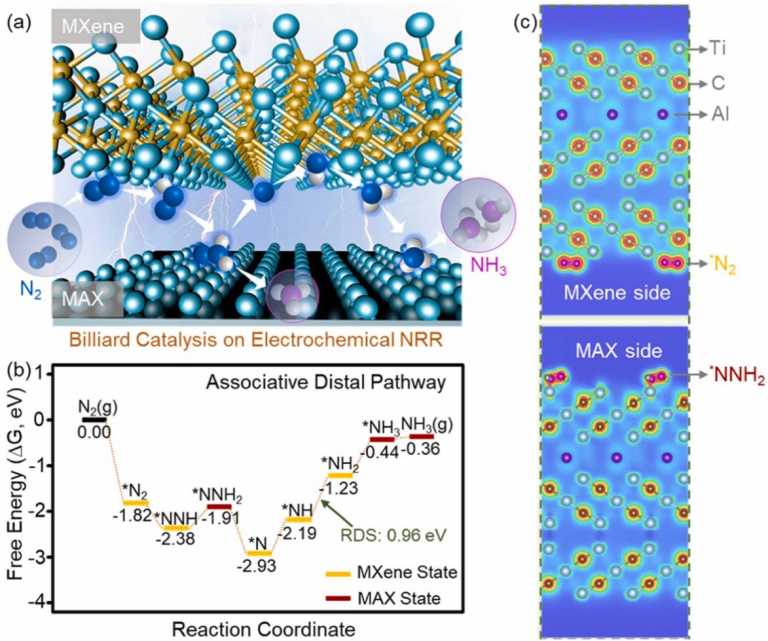
在氮还原反应(NRR)研究中,复旦大学团队构建 Ti₃C₂ MXene/MAX 异质结,通过调控刻蚀程度优化 Al 含量,实现高效电催化合成氨,其 DFT 分析显示,异质结界面处 NH→NH₂步骤的能垒为 0.96 eV,遵循质子 – 电子耦合的末端缔合机制,且 MAX 与 MXene 界面形成 40 mV 表面电位差,促进电子从 MAX 向 MXene 转移,加速 N₂活化。此外,在 Fe 吸附的 N/F/P/S/Cl 掺杂 Ti₃C₂O₂非金属掺杂体系研究中,F 和 S 掺杂显著降低 NNH 形成(-0.20 V)和 NH₃脱附(-0.45 V)的吉布斯自由能,且 Fe 原子向 N₂分子转移电子,削弱 N≡N 键,促进氮气活化,展现出对 NRR 的积极影响。
DOI:https://doi.org/10.1016/j.apcatb.2022.121755
美国普渡大学 Babak Anasori 团队 2025 年发表的关于 W₂TiC₂Tₓ MXene 的 HER 研究论文中,通过 DFT 计算设计,先计算形成能筛选出非 MAX 前驱体 (W,Ti)₄C₄-y 并预测其可刻蚀性,再优化 W₂TiC₂Tₓ的晶体结构,确定表面 – OH 和 – F 终止基团的稳定性;在活性位点与电子结构方面,计算得出 W 顶位和 O 空位对 H的吸附能分别为 – 1.55 eV 和 – 0.77 eV,确认空位可优化活性,DOS 分析显示 W-d 轨道贡献主要近费米能级态密度,促进了 H吸附动力学;性能预测与验证上,理论预测过电位为 144 mV(10 mA/cm²),实验测得值为 148 mV,误差 ,且电荷转移电阻(Rct)计算与电化学阻抗谱(EIS)结果一致,验证了模型可靠性。
DOI:https://doi.org/10.26434/chemrxiv-2024-dprbn
二、DFT理论计算分析方法解析
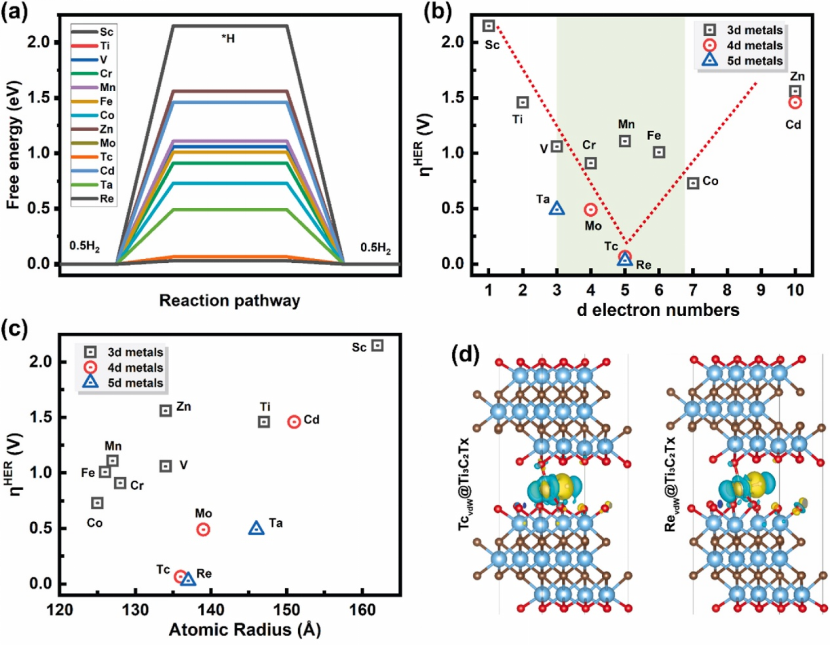
吉布斯自由能计算在电催化反应中具有重要应用,主要用于评估反应路径中各步骤的热力学可行性。例如在氢析出反应(HER)中,可通过计算 H 吸附自由能(ΔG_H)来分析氢吸附过程的难易程度;在氮还原反应(NRR)中,则能用于计算 N₂→N₂H 步骤的能垒,从而判断氮气活化的热力学可行性。其关键参数在于,当 ΔG 接近 0 eV 时,意味着吸附 / 脱附过程接近热力学平衡状态,此时催化剂活性最优。这是因为 ΔG 值趋近于 0 时,反应中间体既不会过强吸附在催化剂表面难以脱附,也不会过弱吸附导致反应难以发生,恰好处于有利于催化反应进行的状态。例如,贵金属 Pt 作为 HER 的基准催化剂,其 H 吸附自由能(ΔG_H*)约为 0 eV,这一特性使其在催化过程中展现出优异的吸附 / 脱附动力学,成为衡量其他催化剂活性的重要参考标准。
DOI:https://doi.org/10.1016/j.jpcs.2025.112635
电子结构分析是揭示催化剂活性位点特性及反应机制的关键手段,主要通过态密度(DOS)和电荷差分图展开。态密度分析聚焦于 MXene 及其复合物的 d 带中心位置,以此阐释活性位点的电子特性,例如在 W₂TiC₂Tₓ MXene 的研究中,其 W 原子的 d 轨道在费米能级附近呈现高密度分布,这种电子结构特征能够有效增强催化剂对 H * 的吸附能力,从而优化氢析出反应(HER)的动力学过程。电荷差分图则通过可视化技术直观呈现吸附过程中电荷的转移方向,为理解催化反应中的电子相互作用提供微观视角,如在 Fe 吸附的 Ti₃C₂O₂体系中,电荷差分图清晰显示 Fe 原子向 N₂分子转移电子,这一过程削弱了 N≡N 键的强度,促进了氮气在氮还原反应(NRR)中的活化。二者结合能够从电子能级分布和电荷迁移规律两方面深入解析催化剂的活性起源,为合理设计高性能电催化材料提供理论依据。
DOI:https://doi.org/10.1016/j.mssp.2024.109091
表面吸附与活化机制是电催化反应研究的核心环节,通过吸附能计算与过渡态搜索可深入解析反应物在 MXene 表面的作用行为。吸附能计算能够量化 N₂、CO₂等反应物在 MXene 表面的吸附强度,为活性位点设计提供关键依据,例如 Ti₃C₂O₂-Fe 体系中 N₂的吸附能为 – 0.85 eV,表明该表面对氮气具有强吸附作用,有利于后续活化过程。过渡态搜索则聚焦于确定反应路径中的过渡态结构,通过计算活化能垒揭示反应动力学瓶颈,如在二氧化碳还原反应(CO2RR)中,通过分析 CO→COOH 步骤的能垒,可明确 C-O 键断裂与重组的难易程度,进而指导催化剂优化。二者结合能够从热力学吸附强度与动力学能垒调控两方面,系统阐释反应物在 MXene 表面的活化机制,为设计具有高效吸附与催化转化能力的电催化剂提供理论支撑,推动电催化反应在能源转化领域的实际应用。

DOI:https://www.cjcatal.com/CN/10.1016/S1872-2067(23)64501-2
三、总结
MXene材料通过密度泛函理论(DFT)计算在电催化领域的研究已形成系统性框架,其应用涵盖活性位点精准设计、反应路径动态优化及催化性能预测等关键层面,为材料理性设计提供了理论支撑。随着计算方法的革新,未来研究将重点推进高通量计算与机器学习协同筛选策略,如乔世璋团队通过机器学习指导MXene基催化剂在C-N偶联反应中的性能突破;同时需发展跨尺度模拟技术,通过量子力学与分子动力学(QM/MD)的深度融合,解析溶剂化效应与电极/电解质界面动态过程;此外,针对MXene在电催化工况下的结构稳定性问题,亟待建立表面官能团动态演化模型,揭示材料在电场、酸性/碱性环境中的构效关系演变规律。这些方法论创新不仅深化了MXene催化机制的基础认知,更为其在电解水制氢、二氧化碳还原等能源转换技术中的实际应用构建了完备的理论体系,推动二维材料催化研究从理论预测向工程化设计跨越。
找华算做计算?专业靠谱省心又省时!
益于理论计算化学的快速发展,计算模拟在纳米材料研究中的运用日益广泛而深入。科研领域已经逐步形成了“精准制备-理论模拟-先进表征”的研究模式,而正是这种实验和计算模拟的联合佐证,更加增添了论文的可靠性和严谨性,往往能够得到更广泛的认可。
华算科技已向国内外1000多家高校/科研单位提供了超过50000项理论计算和测试表征服务,部分计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。
