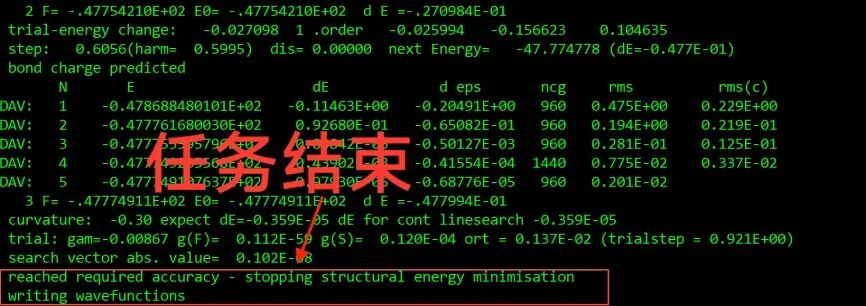
结构优化的核心目标是通过调整原子位置或晶格参数,使体系的总能量达到最小值。在VASP中,这一过程通常通过迭代算法实现,例如共轭梯度法(Conjugate Gradient)或准牛顿法(Quasi-Newton)。优化过程中,系统会根据当前的原子力和能量变化进行调整,直到满足预设的收敛条件(如能量变化小于设定值)。
结构优化的理论基础是密度泛函理论(DFT),其核心是通过交换-相关泛函(如GGA或LSDA)来描述电子结构。在VASP中,常用的交换-相关泛函包括Perdew-Wang 1991(PW91)和PBE等。这些泛函通过近似描述电子之间的相互作用,从而计算体系的总能量和结构。
VASP计算需要准备超算连接软件EASYCONNECT与SSH,建模软件VESTA,超算连接软件Winscp
jp-minerals.org/vesta/en/download.html
EasyConnect下载-EasyConnect最新版下载V7.6.7.0
Downloading WinSCP-6.5.3-Setup.exe :: WinSCP
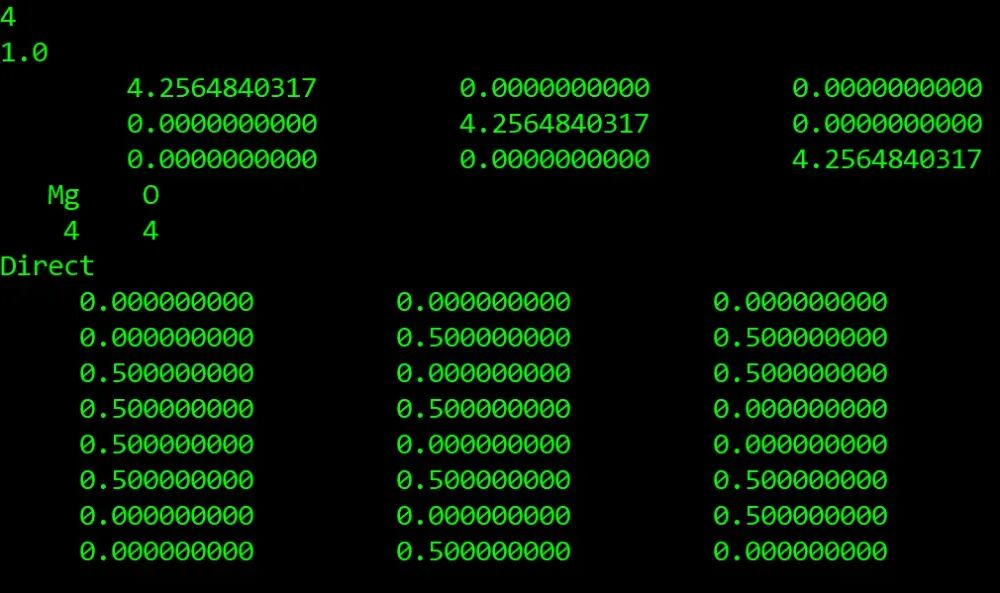
cd 202511/mgo/opt 进入计算文件夹
ISTART=0 #开始新的任务,随机产生初始波函数
ICHARG=2 #开始新的任务,从原子电荷密度产生体系初始电荷密度
PREC=M #计算精度,决定ENCUT,FFT的网格大小
EDIFF=1E-5 #相邻两步电子迭代的能量差收敛标准
EDIFFG=-0.1 #离子弛豫的force的收敛标准
ISMEAR=0 #费米能级附近电子占据数为高斯分布,适合金属、半导体、绝缘体
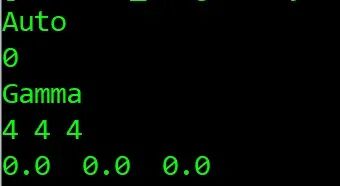
cat KPOINTS 查看KPOINTS文件内容
Gamma #布里渊区K点网格以Gamma点为中心
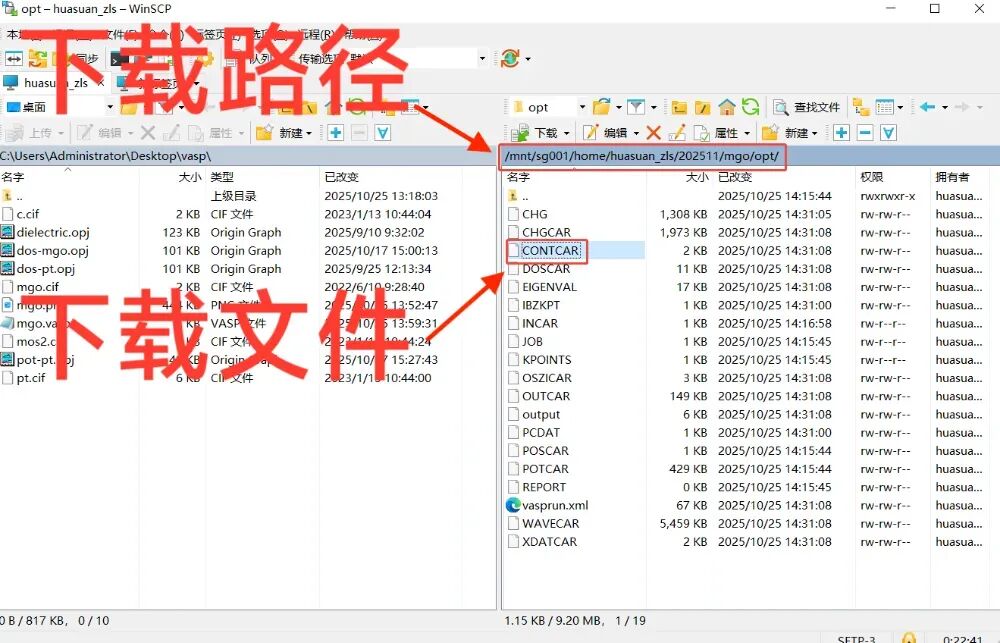
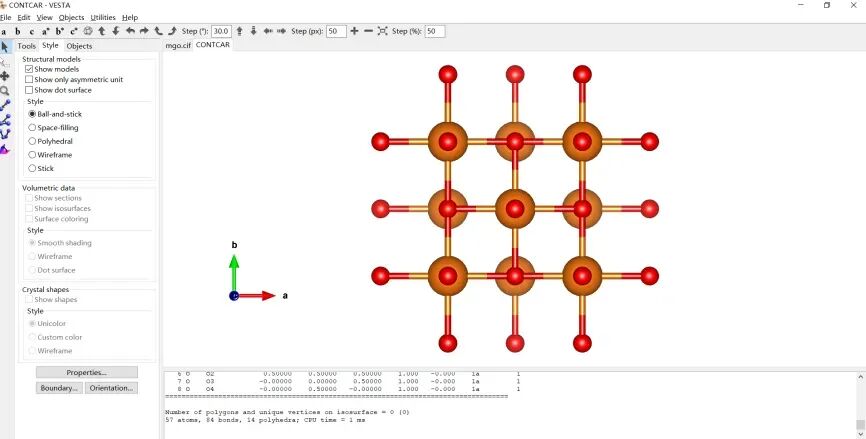
用winscp下载CONTCAR文件,并用VESTA作图
通过本文的学习,大家已经能够初步掌握VASP结构优化的完整流程,包括理解INCAR、POSCAR、KPOINTS等核心输入文件的意义,并独立完成计算任务的配置与提交。这为后续开展更复杂的材料模拟研究奠定了坚实的基础。
【做计算 找华算】
? 华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服务。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!