

Q1:朱老师,请问我这个过渡态计算最有可能是过渡态的是哪个点啊
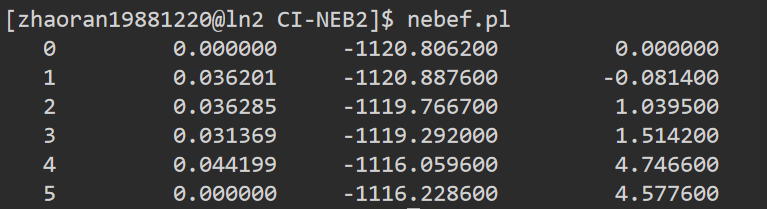

A:如果结构没问题,能量最高点是过渡态
Q2:朱老师,我是要按照这里显示的去下载对应版本的python吗?但我下载好之后 打开还是默认2.7.5版本的
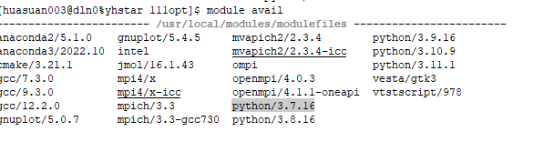
A:加载anaconda3
Q3:朱老师,请问我算出来的DOS为什么没有数据呢?
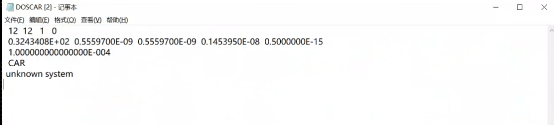
A:电荷文件读取失败,重新复制一下
Q4:朱老师,如果想考虑二维材料的晶界建模要怎么弄呢?
A:用异质结构
Q5:朱老师,请问VESTA如何得到背景是透明的图呢?
A:导出的时候有个选项,make the background transparent
Q6:朱老师,请问我的体系是CoMoS体系,扩胞之后算的总态密度,然后算了Mo原子的d轨道,Co的p轨道发现贡献非常小,想问这是由于扩胞的影响吗?应该不会这么小的
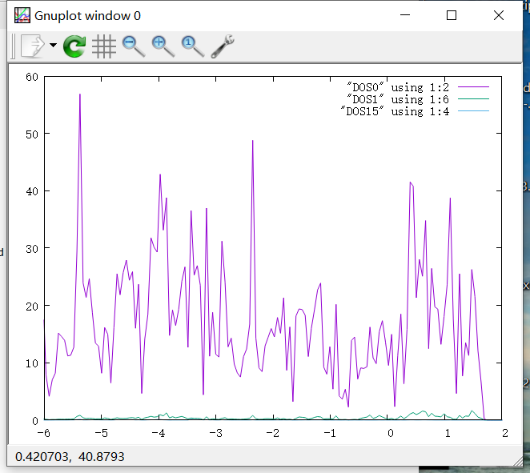
A:分态密度不要和总的比较,和分的比较
Q7:朱老师,我在回复审稿意见的时候遇到一个问题,审稿人说我用这个公式来考虑pH不正确,但是我看很多高水平的文章都是用的这个公式计算自由能啊,我找了好多都没找到说这个公式有问题的文献,我该怎么回这个问题呢?

A:这是经典公式,人家也提了。更时髦的要加入离子,用分子动力学,那个会很麻烦,实在不行引用文献讨论一下了
Q8:朱老师,请问利用COHPCAR.lobster画图时,energy一列的0是不是对应于费米能级呢?
A:是的
Q9:朱老师,我们计算的这个被审稿人说整个PDOS加起来后远远低于TDOS,请问这个是什么参数设置错误的问题吗?
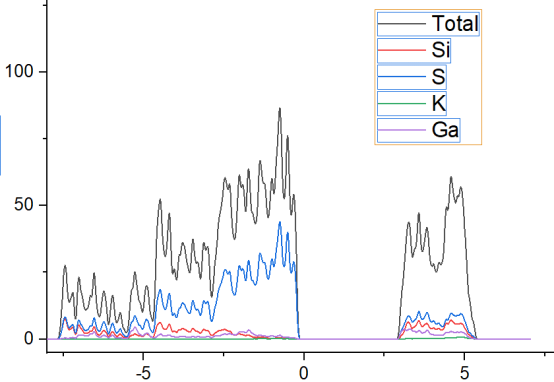
A:正常的,vasp平面波是有这个问题,分太密度强度加起来小于总的,这也不是什么大问题
Q10:朱老师,请问表面态密度图中费米能级以下的表面态是不是就是n型表面态
A:这个是p型,两者都是p
【做计算 找华算】
华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服务。
500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。