

说明:本文华算科技讲解氧化态升高时 XPS 峰向高结合能方向右移的规律,从峰位移本质、原理、典型案例展开,还提及其他影响峰位移的因素。阅读后能掌握 XPS 谱图解读核心依据,理解氧化态与峰位移的关联。
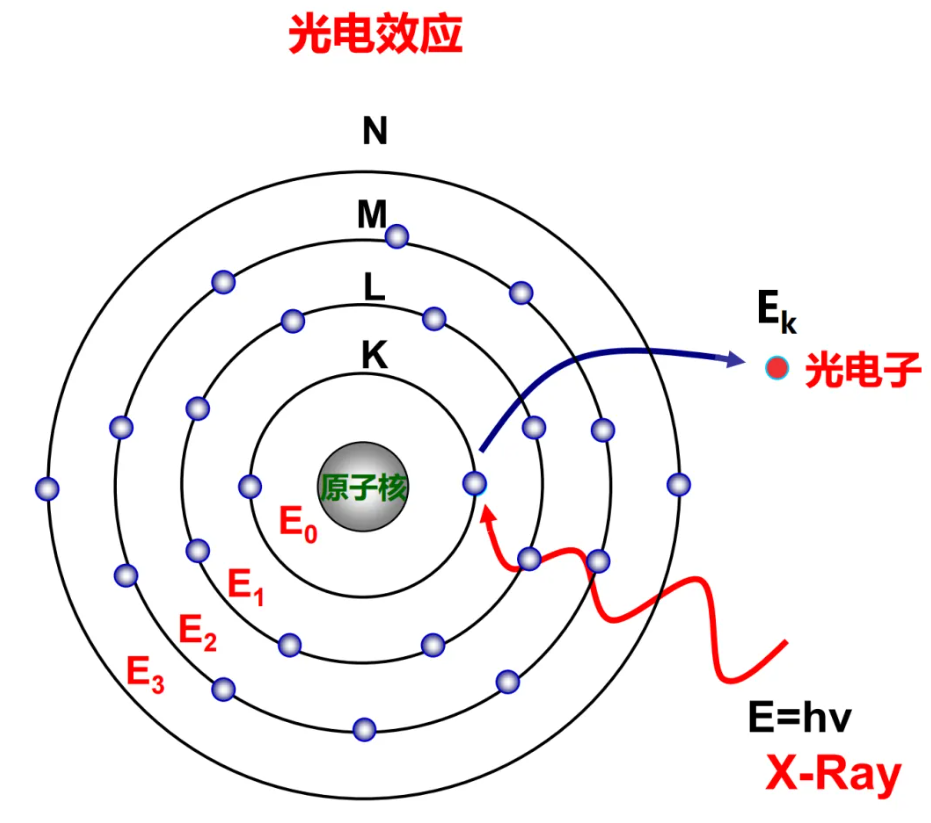
XPS技术的工作原理基于光电效应:用已知能量的X射线照射超高真空环境中的样品,激发原子内层或价层电子成为光电子,通过测量光电子的动能,可推算出核心电子的结合能。

DOI: 10.1021/acsbiomaterials.7b00040
对于同一种元素,其核心电子的结合能并非固定值——当原子所处化学环境改变时,结合能会发生微小变化(通常为零点几到几个电子伏特),这种变化在谱图上就表现为特征峰位置的移动,即峰位移。
其中,向更高结合能方向的移动称为右移,向更低结合能方向的移动称为左移。
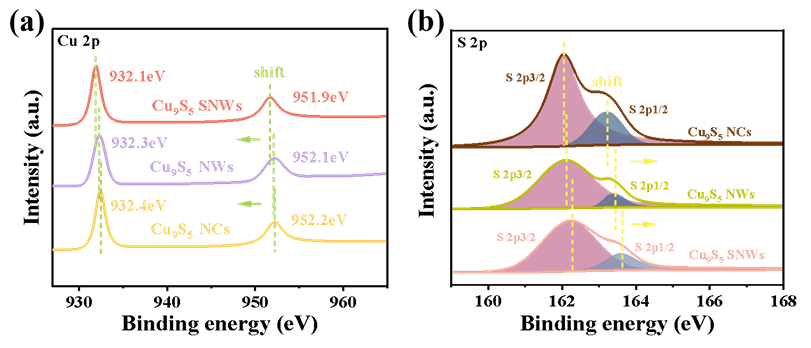
DOI:10.1002/anie.202403023
峰位移的本质是原子局部电子环境的改变。
原子核带正电,核心电子受到原子核的库仑引力束缚,而价层电子和周围原子的电子云会对核心电子产生“屏蔽效应”——电子云密度越高,屏蔽效应越强,核心电子受到的原子核引力就越弱,所需激发能量(结合能)就越低;反之,电子云密度越低,屏蔽效应越弱,结合能就越高。
这一逻辑是理解所有峰位移现象的基础,也是氧化态影响峰位移的核心原理。
明确了峰位移的本质后,氧化态与峰位移的关系为:当原子氧化态升高时,XPS峰一定向高结合能方向(右移)移动。这一结论并非实验现象的简单归纳,而是由氧化态变化引发的电子结构改变所决定的,具体可从两个关键角度理解:
氧化态升高=电子云密度降低
氧化态表征元素在化合物中的得失电子状态,氧化态升高意味着原子失去电子(或成键电子云显著偏离自身),核外电子云密度会净减少。
以金属元素为例,0 价金属单质中原子价电子层完整,电子云密度高且对核心电子的屏蔽效应强,核心电子结合能较低;
当被氧化为 + 2 价阳离子时,原子失去 2 个价电子,电子云密度大幅下降,屏蔽效应减弱,原子核对核心电子的库仑引力增强,激发核心电子成为光电子需更高能量,反映在 XPS 谱图上即峰位向高结合能方向右移。
如金属 Ti(0 价)的 Ti 2p 峰结合能显著低于 TiO(Ti²⁺),而 TiO 的峰位又低于 TiO₂(Ti⁴⁺),清晰呈现 “氧化态升高→峰右移” 的线性关系。
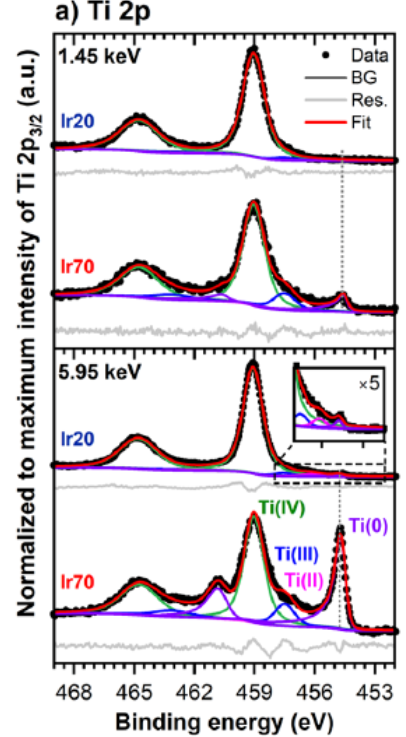
DOI:10.1021/acscatal.3c02948
电子云密度与结合能的直接关联
XPS 峰位的本质是核心电子的结合能,且结合能与电子云密度呈负相关,即电子云密度越低,结合能越高,峰位越靠右;电子云密度越高,结合能越低,峰位越靠左。氧化态升高引发的电子流失,会直接打破原子原有的电子平衡,使核心电子处于更强的原子核引力场中。
这种电子环境的改变具有根本性,因此峰位移方向具备确定性:无论其他因素如何影响,氧化态升高必然伴随峰位右移,这一规律经无数材料表征实验证实,已成为 XPS 谱图解读中最基础、最可靠的判断依据之一。
XPS 分析显示,改性材料中 Mo 3d 与 S 2p 的结合能降低 0.6−0.8 eV,证实 W/O 的掺入提升了材料的电子云密度,且其 O 1s 峰位对应 Mo−O/W−O 键;拉曼光谱中 E₁₂g/A₁g 峰出现红移展宽现象,与 HRTEM 表征结果及理论计算结论一致,共同验证了材料局部结构无序与晶格软化的特征。
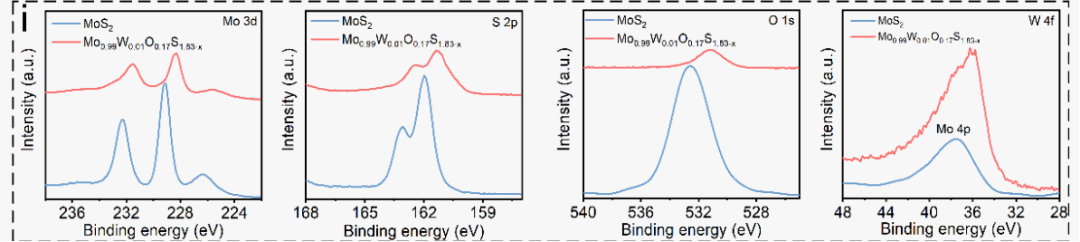
DOI:10.1021/acsnano.5c07443
过渡金属氧化物体系
过渡金属具有多变的氧化态,是研究氧化态与峰位移关系的理想对象。以钴(Co)的氧化物为例:在Co单质(Co⁰)中,Co 2p₃/₂峰的结合能约为778.5 eV;当形成CoO(Co²⁺)时,峰位右移至约780.0 eV;进一步氧化为Co₂O₃(Co³⁺)时,峰位继续右移至约781.5 eV。
这一现象的本质是:随着氧化态从0升高到+3,Co原子逐步失去价电子,电子云密度持续降低,核心电子受到的屏蔽效应不断减弱,结合能逐步增大。在实际科研中,研究者常通过Co 2p峰的位移程度,定量分析样品中Co²⁺与Co³⁺的比例,为催化反应机理研究提供关键依据。
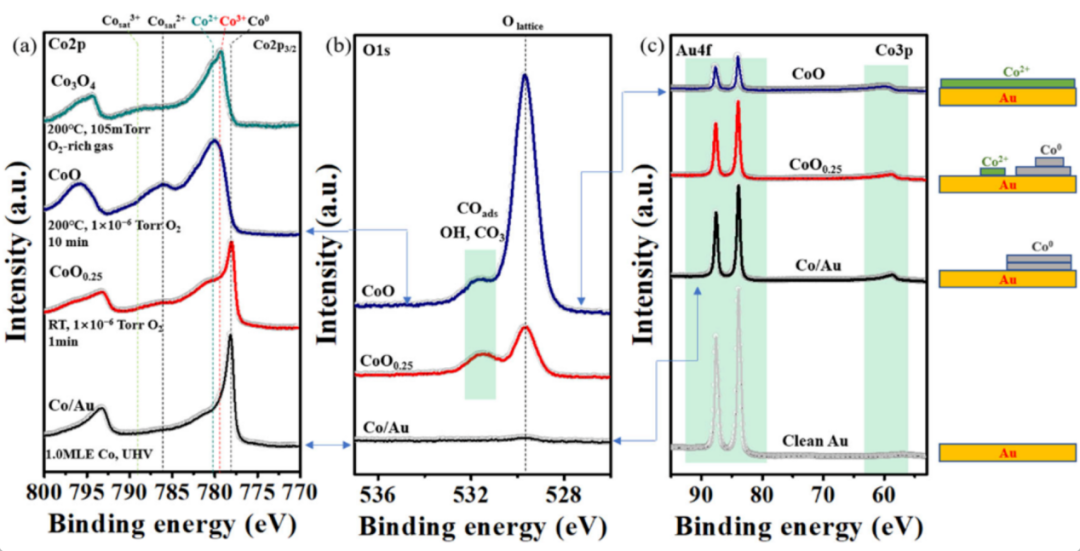
DOI:10.1038/s41467-023-42301-7
半导体掺杂体系
X射线光电子能谱测试中N(1s)谱图显示,经过紫外线照射的FBDPPV/iPAD-1薄膜在402 eV处出现四价氮信号,证明PAD-1⁺阳离子的生成。
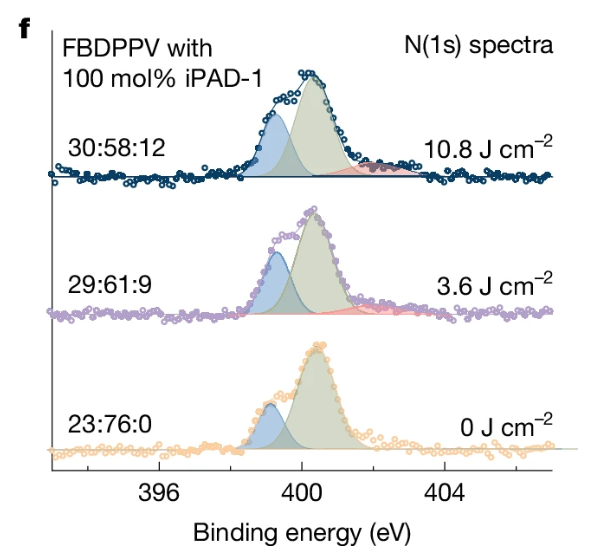
DOI:10.1038/s41586-025-09075-y
氧化还原体系
LM-LCO 表面存在 La、Mo 元素且具有 3d 自旋-轨道双重态结构,证实LMO 改性后 LM-LCO 中 Co²⁺/Co³⁺比值无明显变化,说明 LMO 层未改变 Co 的价态;O 1s显示 LM-LCO 表面氧空位更多,这归因于其表面 LMO 层含大量氧缺陷。
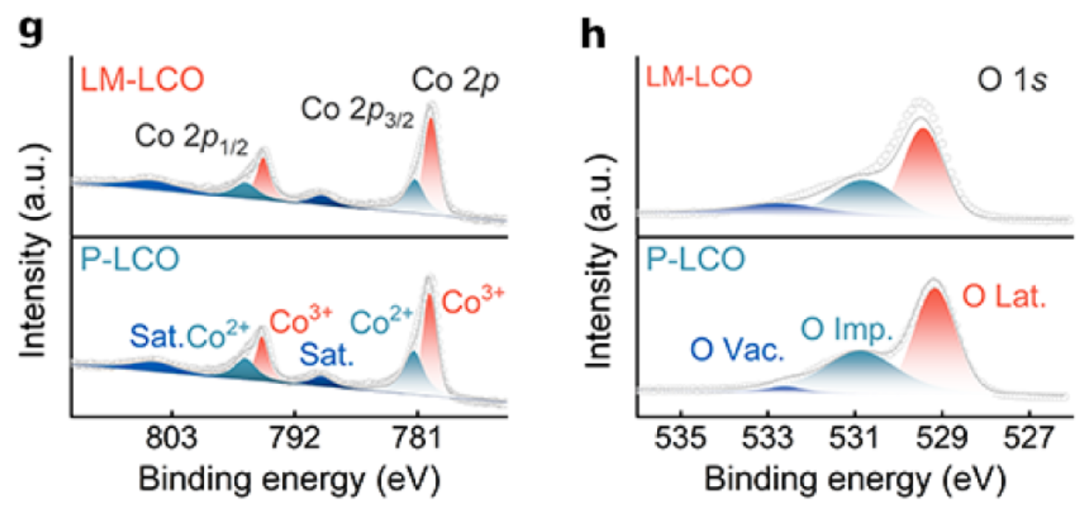
DOI:10.1021/acsnano.5c06237
需要强调的是,XPS峰位移是多种因素共同作用的结果,氧化态只是其中最核心的因素。在实际谱图解读中,还需考虑以下因素,避免单一依据氧化态判断峰位移方向导致的误判:
电子云密度的直接改变(非氧化态因素)
除了氧化态变化,原子与不同电负性元素成键也会直接改变电子云密度。当中心原子与电负性更强的原子成键时,成键电子云会向电负性强的原子偏移,导致中心原子电子云密度降低,结合能升高,峰位右移——这一现象与氧化态升高的效果类似,但中心原子的形式氧化态并未改变。
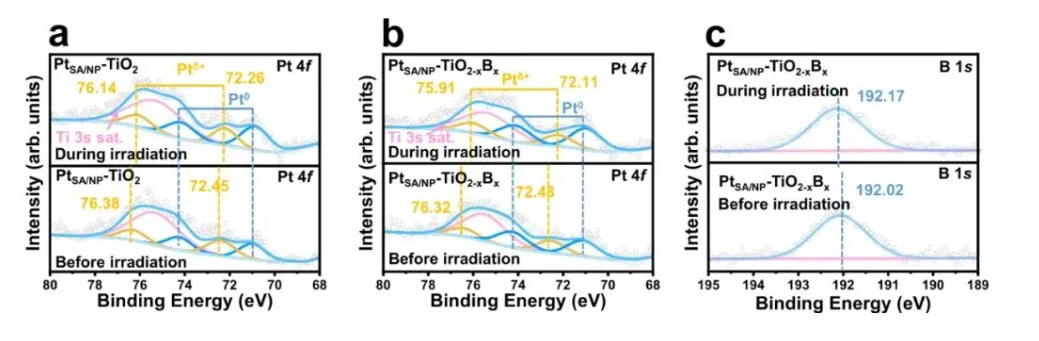
DOI: 10.1038/s41467-025-63637-2
表面态变化:缺陷与吸附
XPS具有极高的表面敏感性,样品表面的缺陷(如氧空位)和吸附物种会显著改变表面原子的电子环境,进而影响峰位。
表面缺陷:在金属氧化物表面,氧空位的形成会留下2个电子,这些电子会局域在邻近的金属阳离子上,使其还原为较低价态(氧化态降低),导致峰位左移。
如图 O 1s 谱中约 532 eV 的峰常被认为与氧空位相关,但该峰并非氧空位独有,O22-、吸附羟基等物种也有类似结合能,且环境湿度会影响吸附羟基含量,可能误导氧空位含量判断。
氧空位产生时的电荷补偿会导致金属还原形成低价金属离子,可通过检测金属的 XPS 信号辅助验证氧空位。
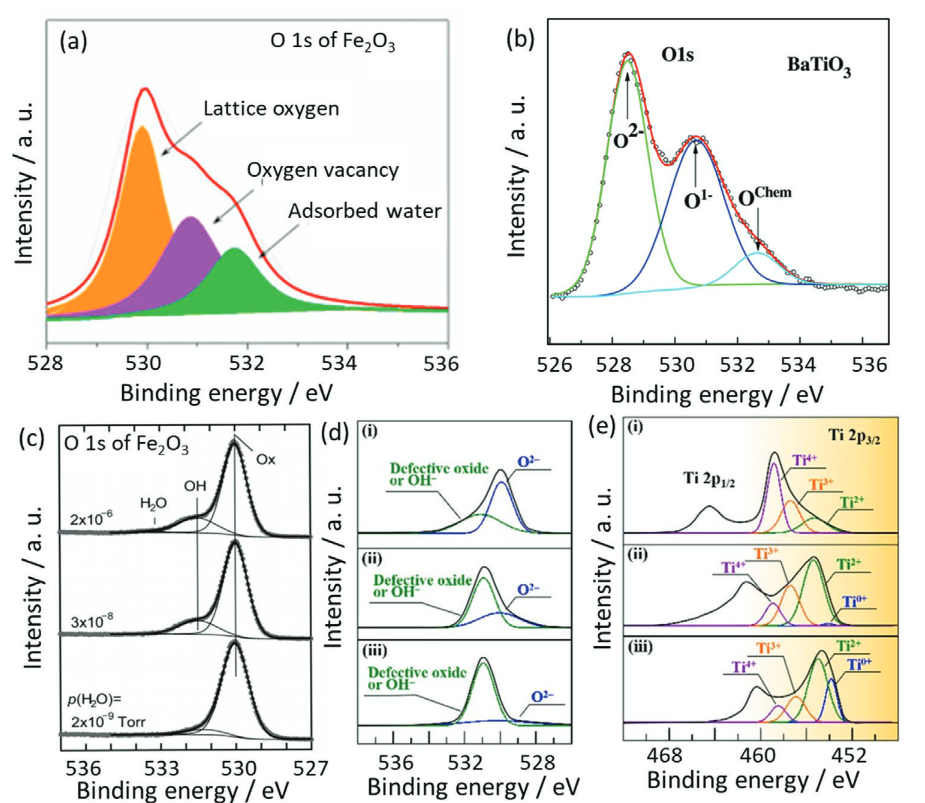
DOI: 10.1002/adfm.202109503.
电荷转移过程
电荷转移是电子在不同原子、分子或材料区域间的转移,会直接改变相关原子的局部电荷状态。异质界面处,因功函数或费米能级差异,电子会自发转移:电子从表层原子转移至衬底时,表层原子电子云密度降低,XPS 峰位右移;反之则左移。
如图采用原位辐射 XPS 研究 CdS/BiOBr 异质结界面电荷转移行为发现,异质结形成后,Cd 3d(404.9eV、411.7eV)与 S 2s(225.8eV)结合能均向高能方向移动(Δ≈0.3eV),直接证实电子从 CdS 向 BiOBr 自发转移。
BiOBr 的 Bi 4f、Br 3d 及晶格氧 O 1s 峰呈现完全相反的变化趋势,这种对称性结合能位移为 S 型异质结电荷转移机制提供了证据。
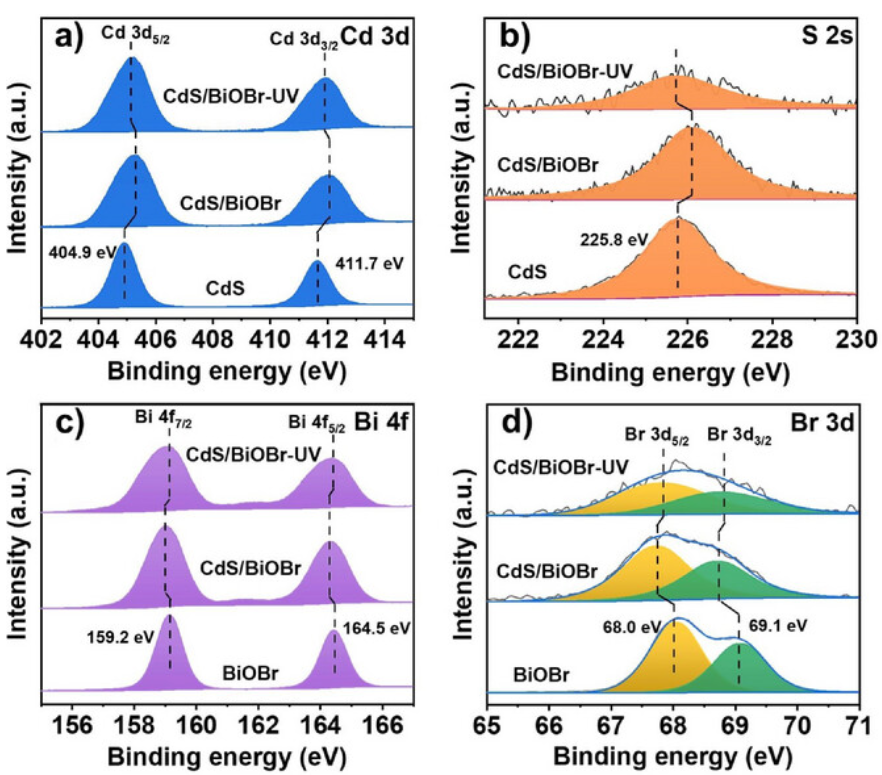
DOI:10.1002/anie.202505456
【高端测试 找华算】
华算科技是专业的科研解决方案服务商,精于高端测试。拥有10余年球差电镜拍摄经验与同步辐射三代光源全球机时,500+博士/博士后团队护航,保质保量!
?已助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果在Nature&Science正刊及子刊、Angew、AFM、JACS等顶级期刊发表!