

所谓电子自旋调控催化,是指通过操控电子的自旋状态(如自旋极化、自旋翻转、自旋选择性传输等)来影响催化剂表面吸附、活化与反应路径的方式。这种研究视角不仅扩展了催化科学的理论框架,也为设计新型高效、选择性的催化材料提供了新的维度。
从理论计算的角度来看,电子自旋的作用在基于密度泛函理论(DFT)的自洽计算中得以清晰呈现。自旋相关的交换-关联泛函、费米能级处的自旋态密度分布、以及磁性有序结构对表面电子态的调控,均能够直接通过计算得到,并进一步与反应能垒、过渡态结构以及催化活性进行关联。
相比于传统的“无自旋”处理方式,引入自旋自由度不仅能更准确地再现实验观测结果,还能揭示许多仅凭价态和能带结构无法解释的反应规律。因此,从理论层面全面系统地探讨电子自旋调控催化的机理,对于推动这一新兴方向的发展具有重要意义。
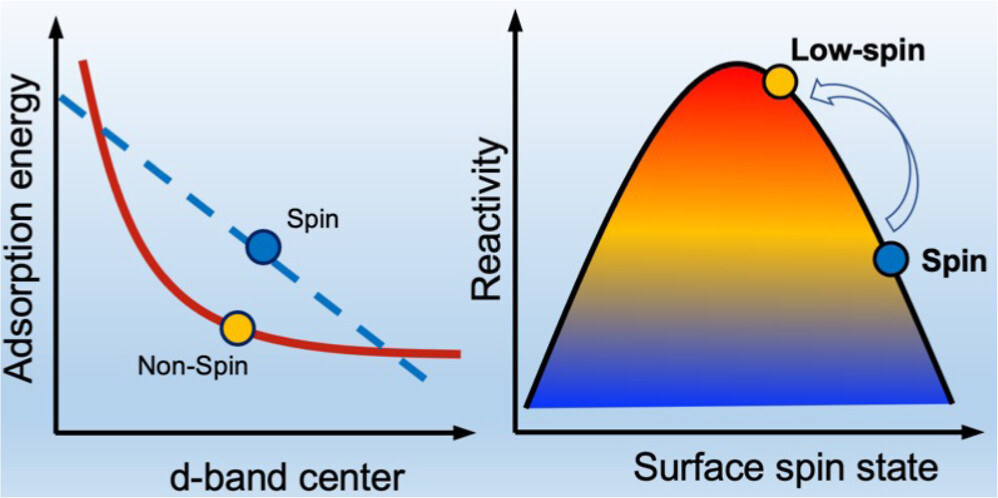

电子自旋与催化反应的基本关系
电子自旋调控催化的核心在于电子态的分裂与自旋选择性对反应过程的影响。在量子力学框架下,电子的自旋不仅是一个基本量子数,还直接决定了电子在外加磁场和交换场作用下的能量差异。
当电子在催化剂表面与吸附物发生相互作用时,其自旋取向往往会影响态密度分布与轨道杂化强度,从而决定反应物分子的活化程度。例如,对于过渡金属催化剂而言,其d轨道在自旋上往往是非对称分布的,即存在明显的自旋极化。正是这种自旋极化效应,使得吸附分子的成键轨道与反键轨道能够选择性地与某一自旋态发生耦合,从而改变化学键的断裂与生成过程。
理论计算中,这种效应最直观的体现是自旋分辨态密度(spin-polarized density of states, DOS)的变化。通过对比自旋向上与自旋向下的电子态,可以发现催化剂表面的能级结构在不同自旋通道中存在差异。这种差异会导致吸附能的不对称,从而为某些反应路径提供额外的热力学或动力学优势。
与传统催化描述不同,自旋的调控使得反应过程不仅依赖于几何结构与能量最低化原则,还受到量子自旋选择规则的约束,从而在反应速率、选择性甚至中间体稳定性上展现出新的规律性。
理论计算方法对自旋调控的刻画
要深入理解电子自旋在催化中的作用,必须依赖精确的理论计算工具。密度泛函理论(DFT)提供了一种可行途径,通过引入自旋密度泛函(spin density functional)来同时处理电子的空间分布与自旋分布。在自旋极化计算中,体系的总电子密度被拆分为自旋向上与自旋向下两部分,并通过交换-相关能泛函进行自洽求解。这样,不同自旋通道的态密度、能带以及反应路径都能够分别计算并比较。
在实际操作中,自旋相关计算的复杂性远高于非自旋情况。
首先,计算需要考虑不同自旋组态的能量差异,尤其是在涉及磁性材料或过渡金属的体系中,自旋排列方式(如铁磁、反铁磁或亚铁磁)会显著影响计算结果。
其次,在过渡态搜索中引入自旋变量,会使得反应能垒对初始自旋态更加敏感,这要求研究者在构建过渡态结构时,必须考虑可能的自旋翻转或自旋交叉。
此外,对于具有强关联效应的体系,还需要结合DFT+U、杂化泛函甚至动态平均场理论(DMFT)等方法,以更准确地再现自旋电子的局域化与离域化特征。这些计算方法的逐步发展,使得电子自旋调控催化的理论研究具备了坚实的基础。
自旋极化与能带结构对催化性能的影响
电子自旋调控催化的显著特征在于其对能带结构与态密度分布的直接调节作用。催化剂的活性往往与其费米能级附近的电子态密切相关,而自旋极化则能够在不同自旋通道中引入能带分裂,进而改变电子填充与能带宽度。例如,在自旋极化的过渡金属表面,d带中心位置在自旋向上与向下通道之间存在差异,这不仅改变了催化剂与吸附分子之间的轨道耦合强度,还可能导致反应能垒的降低或升高。
理论计算结果表明,自旋极化效应在某些关键反应中起到决定性作用。例如,氧分子的解离往往依赖于三重态的自旋结构,若催化剂表面能够提供与之匹配的自旋极化通道,反应速率将显著提升。
另一方面,自旋翻转过程的发生也可能成为反应瓶颈,从而影响整体反应动力学。通过计算不同自旋态下的势能面,可以发现某些反应路径在特定自旋通道中具有更低的过渡态能量,这揭示了自旋选择性催化的基本物理机制。更重要的是,这种效应为能带工程与自旋设计提供了新思路,即通过调控催化剂的磁性与电子结构,主动引导反应物在特定自旋通道中发生转化。
DOI: 10.1039/D4SC04370G
自旋调控催化中的量子力学机制
电子自旋调控催化的深层机理不仅仅停留在能带与态密度的层面,还涉及量子力学中的交换相互作用与自旋选择定律。交换相互作用源于电子自旋对称性对波函数的约束,它决定了不同自旋组态的能量差异。
在催化体系中,交换相互作用往往通过改变反应物与催化剂之间的轨道重叠来调节反应能垒。例如,当反应物分子轨道与催化剂自旋态不匹配时,电子转移过程会受到抑制,导致反应减缓;反之,自旋匹配则会增强电子跃迁,从而加速化学反应。
自旋选择定律在某些反应中起到决定性作用,尤其是涉及自由基或过渡态的过程。根据量子力学的自旋守恒规则,许多反应必须保持总自旋量子数守恒才能发生。这意味着,在自旋调控下,某些本不可能的反应路径可以通过自旋翻转或自旋交叉得以实现,而某些传统路径则可能因自旋不匹配而被禁止。
理论计算在这里的作用是,通过非绝热动力学模拟与自旋相关能量面分析,揭示自旋如何主导反应路径的选择。这种量子层面的描述不仅为实验观测提供了合理解释,也为通过自旋调控设计新型催化剂提供了理论依据。
电子自旋调控催化的前景与挑战
电子自旋调控催化作为一个新兴的研究领域,展现出巨大的应用潜力。从能源转化到环境治理,自旋调控催化为提高反应速率、提升选择性和降低能耗提供了新的可能。然而,从理论计算的角度来看,这一方向仍然面临诸多挑战。
首先,现有的交换-关联泛函在处理强自旋关联与非绝热效应时仍存在一定的不足,如何开发更精准的自旋相关泛函仍是理论发展的关键。
其次,模拟自旋翻转过程需要大规模的非绝热分子动力学计算,这对计算资源与算法效率提出了更高要求。
此外,如何在理论计算中同时考虑外加磁场、电场与自旋轨道耦合效应,也是实现真实模拟不可或缺的步骤。
展望未来,电子自旋调控催化将不仅仅局限于学术探索,而会逐渐走向应用。通过理论计算与实验研究的紧密结合,人们有望实现对催化过程的全方位自旋控制,从而开发出新型高效的电催化剂、光催化剂以及热催化剂。
在这一过程中,理论计算的价值不仅在于解释现象,更在于提供设计原则与预测能力。可以预见,随着计算方法与计算资源的持续进步,电子自旋调控催化将在未来成为催化科学的前沿热点,为能源与环境问题的解决提供新的理论支撑与实践路径。