

Q1:朱老师,我计算出来的H和Li元素态密度有p轨道电子,请问这个该怎么理解呢?
A:这个不用理他,都会有spd的,理论上是0,但是数值计算有时候并不是0,所以一般忽略就可以
Q2:朱老师,请问这里说的结构优化的单gamma点计算是怎么生成的呀,是这里的都改成 1 1 1是吗 ?

A:对的
Q3:朱老师,这个是我氮化镍吸附了一个羟基的吸附模型,但是一跑就散架了 这种情况可能是什么原因啊?
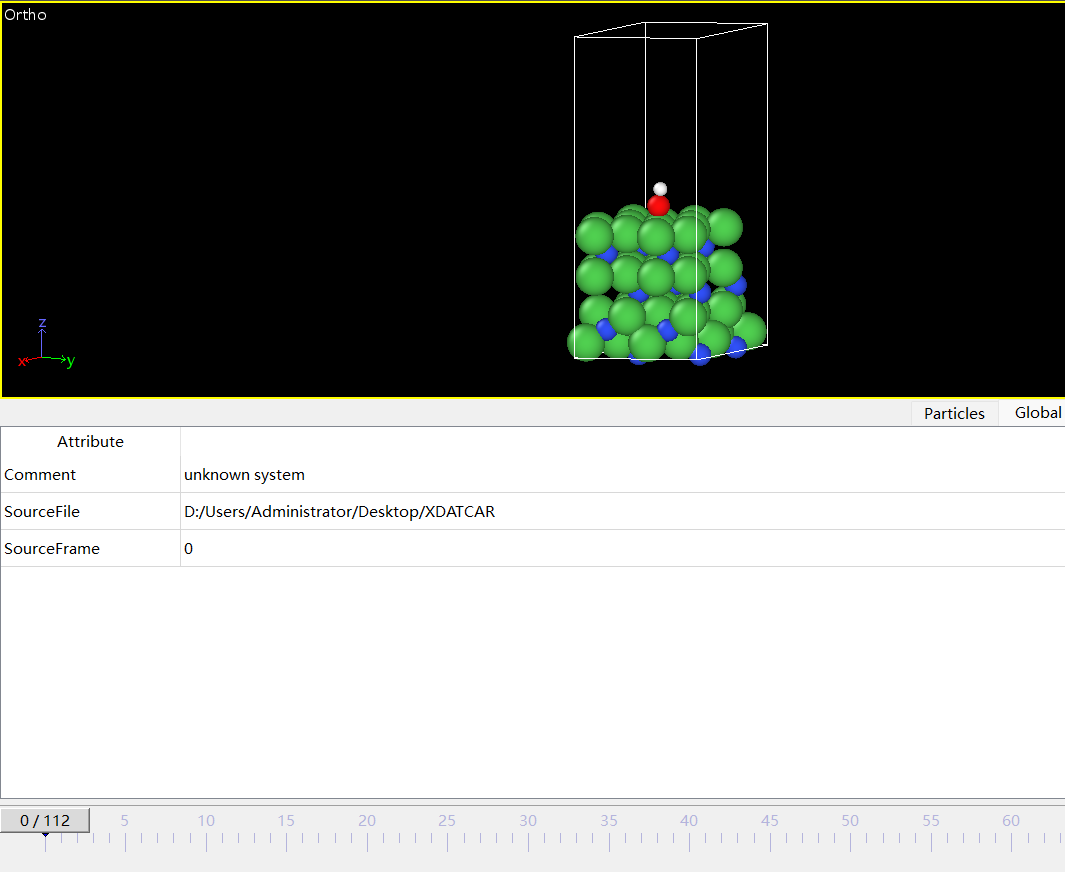
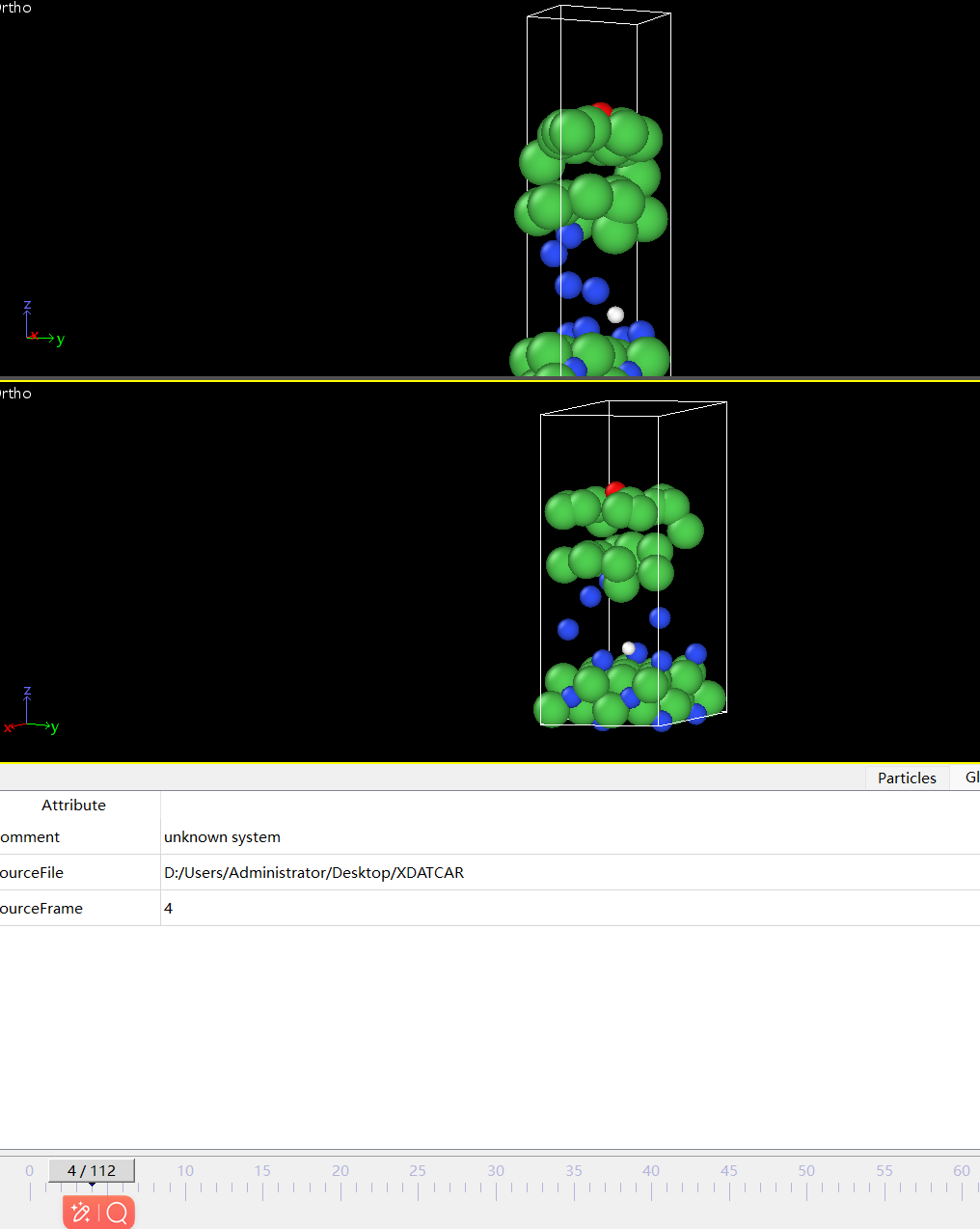
A:可能初始构型不合理,把OH离得远一些
Q4:朱老师,Li吸附在NiO111面上计算出来是这样的结构算是散了吗?
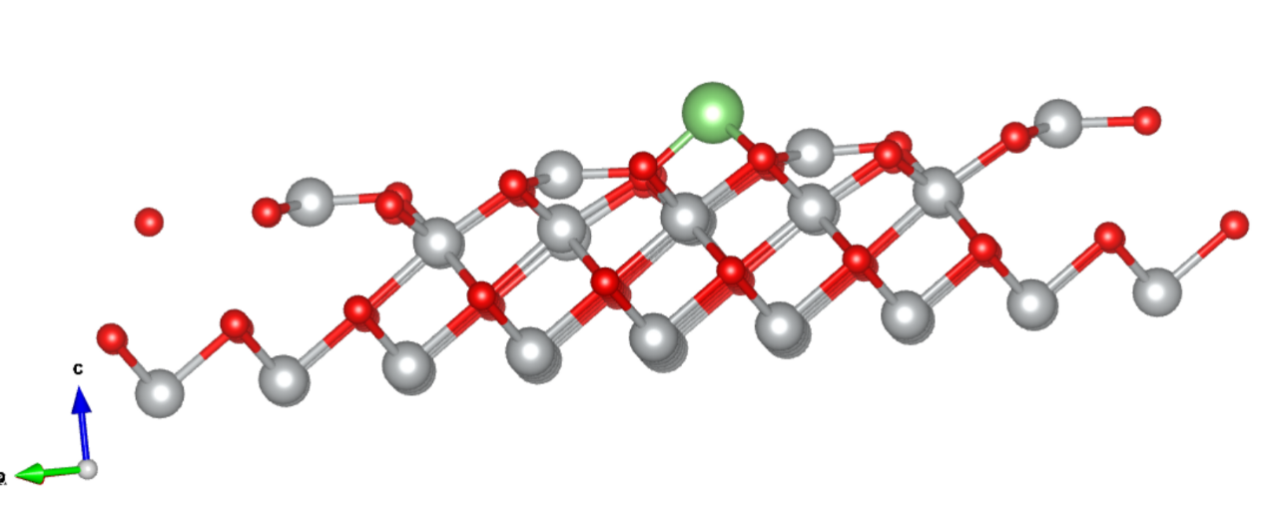
A:没有,这个是周期性边界显示问题
Q5:朱老师,请问一下怎么去设置元素的不同价态呢,通过调节什么参数来实现呢?
A:没办法设置,只能控制总体电荷
Q6:朱老师,掺杂中有200步不收敛的情况吗?
A:正常,加步数就可以
Q7:朱老师,这个PMS在吸附计算的时候S-O键断开了,是什么原因呢?
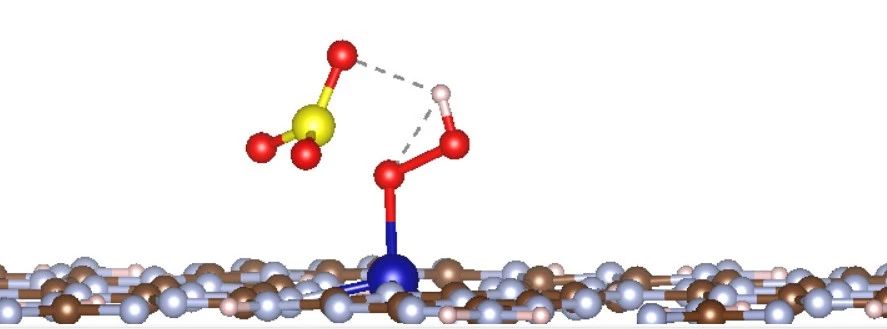
A:这个不算断开,只是一定程度拉长了,正常
Q8:朱老师,我想问一下 如果我想计算二维材料的能带结构 然后用课程上的软件查到的高对称点是只要把带有那些第三列为0的点选上还是说把所有点都选上 但是如果第三列不为0就把那个改为0?
A:用第二种做法,但是避免重复点
Q9:朱老师,如果我想把只有选中部分的电荷密度,从chgcar里面拿出来,该怎么处理呢?
A:做不了,只能截图
Q10:朱老师,做掺杂的时候先掺杂在切晶面还是切完了晶面再做掺杂呢?
A:先切面再掺杂,如果要做整体均匀掺杂就先掺杂在切面,如果只是研究那个杂原子的影响,就先切面,让后把杂原子放在表面
理论计算遇到报错?一键领取《理论计算常见错误答疑汇总》,快速排查VASP/CP2K等计算难题。
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!