

Q1:朱老师您好,我做结构优化,报错信息显示这个,该怎么解决呢?

A:换下ALGO试试
Q2:朱老师,赝势里是不是默认稀土的电子态是基态,有没有处理激发态的办法,比如Sm的激发态和基态就混淆得很厉害
A:所有元素都是基态,没有激发态
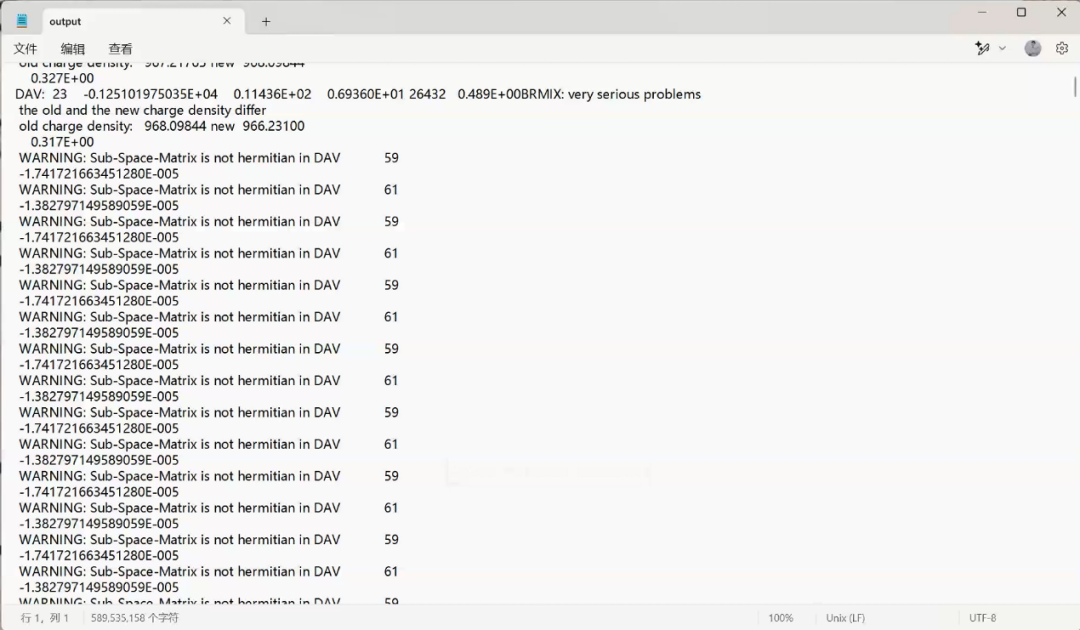
Q3:朱老师,算能带遇到这个问题
A:可能k点太多,内存不够,减少K点
Q4:朱老师,计算Ti3C2的形成能,算3个Ti的能量时,用原胞里Poscar中取掉2个C后剩下的3个Ti,还是算单质Ti的能量乘以3?两种差别很大?
A:单质中原子能量
Q5:朱老师,aimd过程中掉温怎么办?
A:前0.5ps温度有波动正常,截掉这部分就可以
Q6:朱老师,算态密度遇到了这个问题
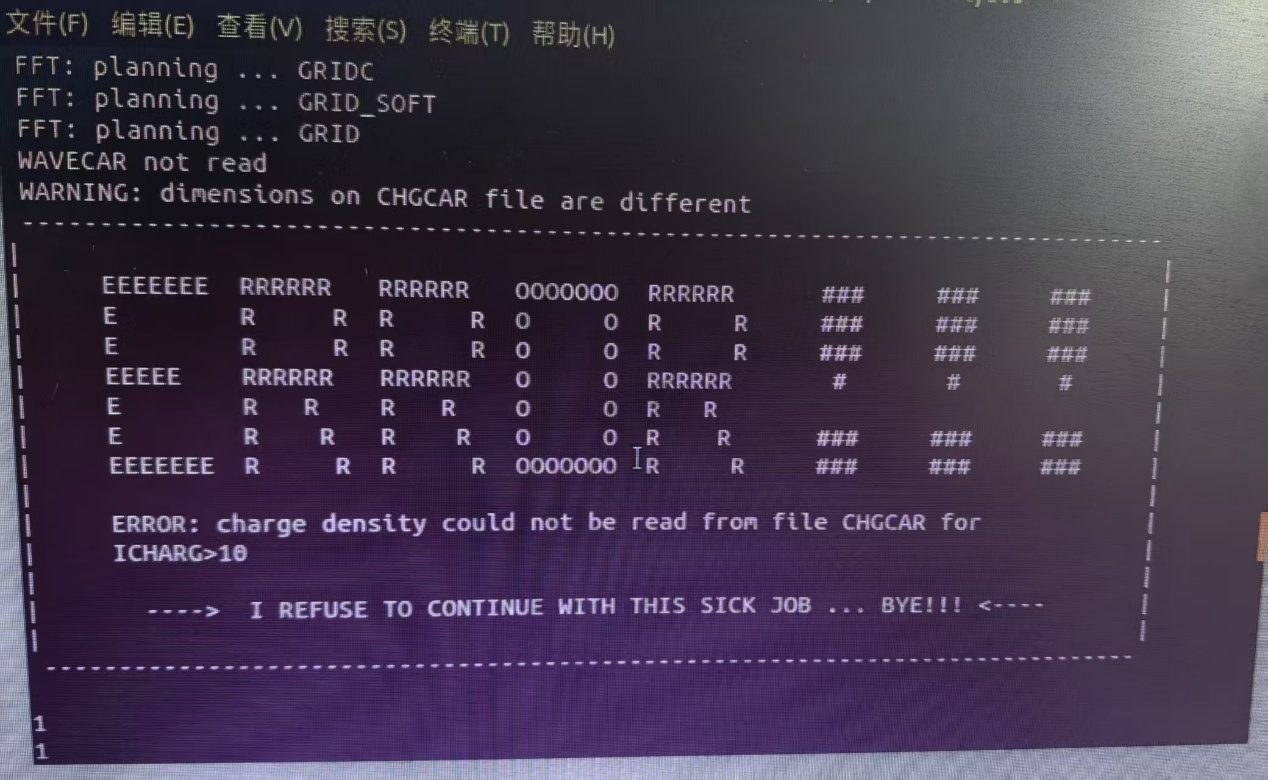
A:重新复制一下CHGCAR WAVECAR,再重算
Q7:朱老师您好,我做slab模型的结构优化,报错如下,请问该如何调整呢?
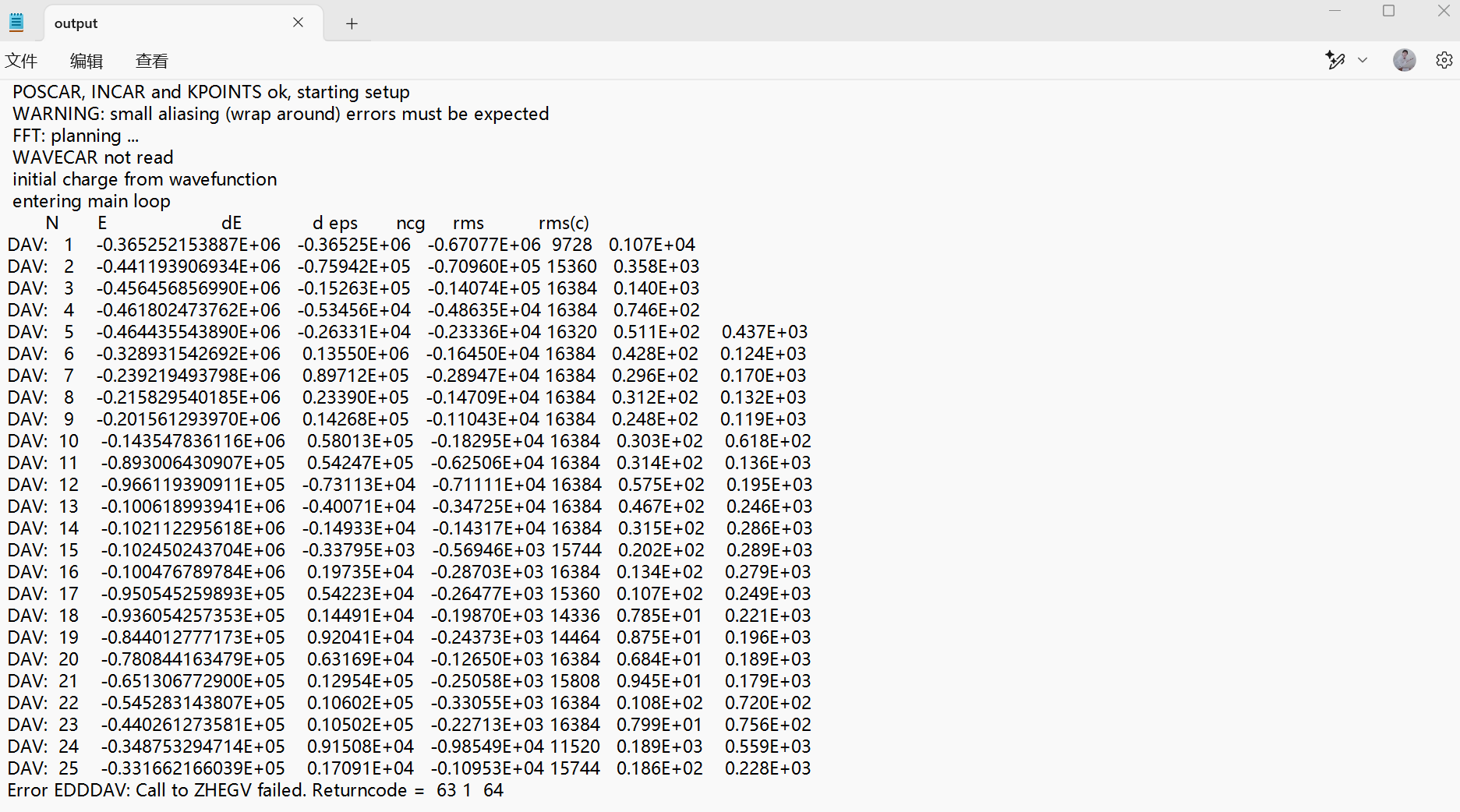
A:加入AMIX=0.1试试
Q8:朱老师,为什么我的静电势算出来只有一边有平台?
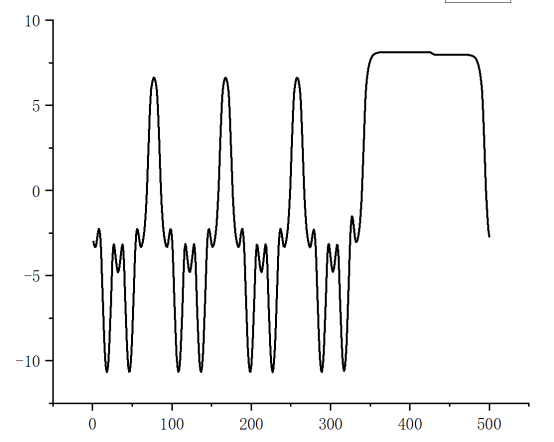
A:和模型位置有关,两个平台在中间有跳跃
Q9:朱老师,一般在计算过渡单金属的吸附 如Co Mo等金属时,需不需要考虑+U呢?
A:涉及到有电子结构性质计算的话可以加
Q10:朱老师,吡啶氮类型的铁单原子催化剂和吡咯氮类型的铁单原子催化剂,通过物理测试手段测试过了它的的磁矩大小数值,想和理论计算的磁矩数据结合起来分析,那是结合理论计算中的outcar中的最后一个mag净磁矩数据分析呢?还是结合投影到铁原子上的那个具体原子磁矩呢?净磁矩的数值貌似比投影大,结合哪个数值分析更好
A:都可以,保持统一就好,投影会损失一部分是正常的
Q11:朱老师,在算过渡态时您的视频里讲到了粗算和精算。我想问下我的正常结构优化收敛标准是EDFIFF = EDIFF = 1E-5, EDIFFG = -0.02.我在粗算和精算时这两个值设多少合适啊?
A:ediff不变,ediffg可以选择0.8-1.0和0.02-0.05
理论计算遇到报错?一键领取《理论计算常见错误答疑汇总》,快速排查VASP/CP2K等计算难题。