

在分子科学与材料物理研究中,弱相互作用(weak interactions)是决定体系结构稳定性与功能特性的关键因素。
与强相互作用(如共价键、离子键、金属键)不同,弱相互作用能量相对较小,通常在0.1–50 kJ/mol范围内,却在自组装结构形成、生物分子识别、分子吸附、表面催化等过程中发挥着决定性作用。
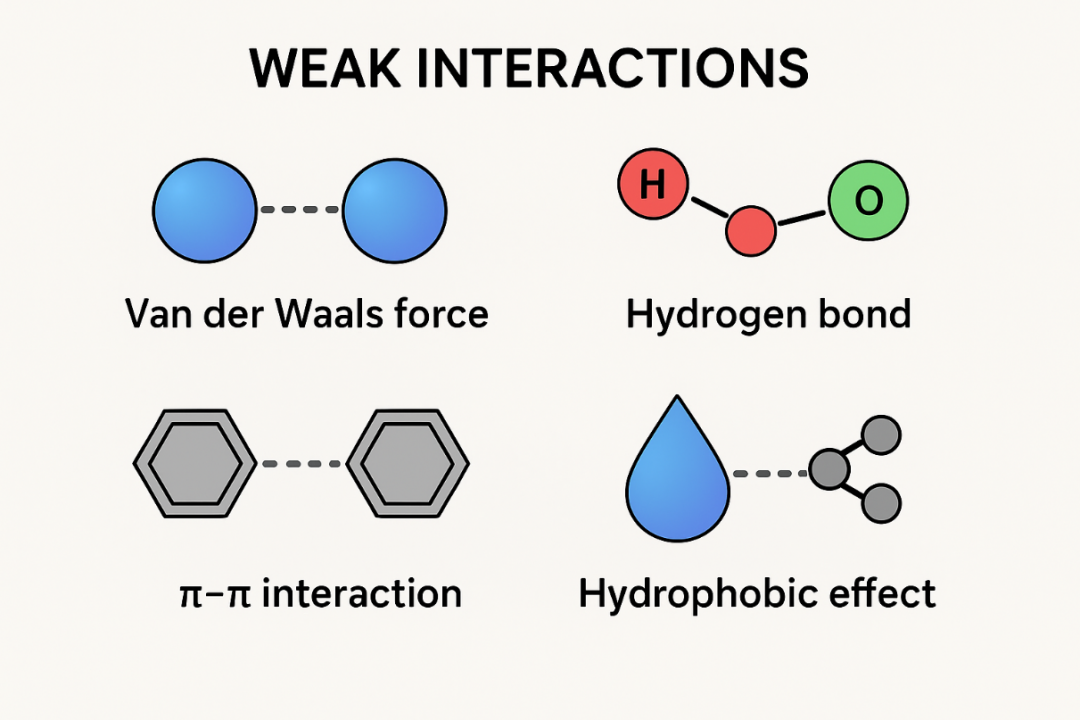
弱相互作用的典型类型包括范德华力、氢键、π–π堆积、疏水作用以及偶极–偶极相互作用等。这些作用虽各具特点,但本质上都源于电子密度分布、静电场相互作用与量子色散效应的综合结果。
由于弱相互作用能量往往较小,其研究需要高精度的理论工具与计算方法支持。实验上弱相互作用难以直接测量,但在理论计算中可通过量子化学与分子模拟手段实现能量分解与空间分布分析。
近年来,随着计算方法的发展,如对称适应微扰理论(SAPT)、能量分解分析(EDA)、独立梯度模型(IGMH)、局域化能量密度(NCI/ELF)等逐渐成为解析弱相互作用的主要方法。
本文华算科技将从弱相互作用的本质出发,系统介绍其计算分析方法,并讨论如何通过不同理论框架深入理解其机理。

理论基础与分类
弱相互作用的理论根源在于分子间或分子内电子云的微妙作用。其最基本的类型是范德华力,包括色散作用与诱导作用。色散作用来源于瞬时偶极–诱导偶极相互作用,是普遍存在的量子效应;诱导作用则源于极性分子诱导非极性分子电子云偏移而产生的吸引。
氢键则是最广为人知的一类弱相互作用,其强度介于典型共价键与范德华力之间,由氢原子与高电负性原子之间形成的部分静电与轨道相互作用综合决定。
除此之外,π–π作用在芳香环堆积、蛋白质结构保持等方面至关重要,疏水作用则常见于溶液中分子自组装过程。
从能量尺度来看,氢键强度通常为5–30 kJ/mol,π–π作用与范德华作用多在0.5–5 kJ/mol,疏水作用强度则依赖环境与分子种类。尽管这些相互作用能量较低,但在多体体系中可能通过协同效应产生巨大影响。
例如DNA双螺旋的稳定性依赖于氢键与碱基堆积的综合作用,而多孔材料中分子吸附与扩散则由范德华作用与静电势共同主导。因此,弱相互作用的研究不仅是学术问题,更是理解自然与设计功能材料的重要基石。
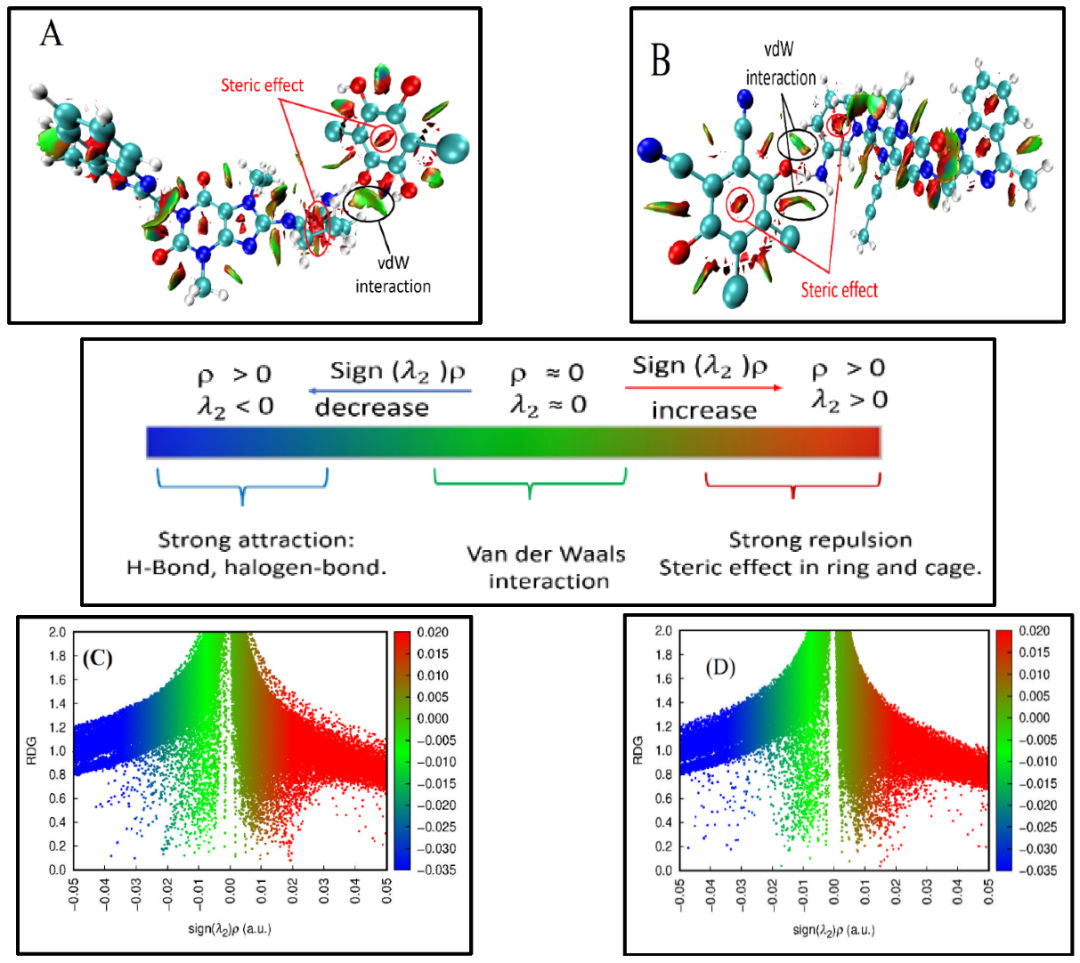
DOI:10.3390/molecules27196320
量子化学方法的应用
量子化学计算是研究弱相互作用最基础且最精确的理论工具之一。传统的密度泛函理论(DFT)在处理共价键问题上表现良好,但对色散相互作用往往存在低估或缺失。
因此,发展了多种修正方法,如DFT-D2/D3色散修正、vdW-DF函数以及MBD(Many-Body Dispersion)方法,以在DFT框架下引入色散效应。这些方法在计算大分子堆积能、表面吸附能时尤为重要。
更高精度的方法包括对称适应微扰理论(SAPT),它能将总相互作用能分解为静电、交换、诱导与色散等分量,为深入理解弱相互作用的来源提供清晰物理图景。
SAPT在分子簇、药物分子–受体体系研究中应用广泛,能区分不同能量成分对总结合能的贡献。除此之外,能量分解分析(EDA)方法亦常用于配合物与超分子体系中,通过量化各能量项揭示弱相互作用的本质。
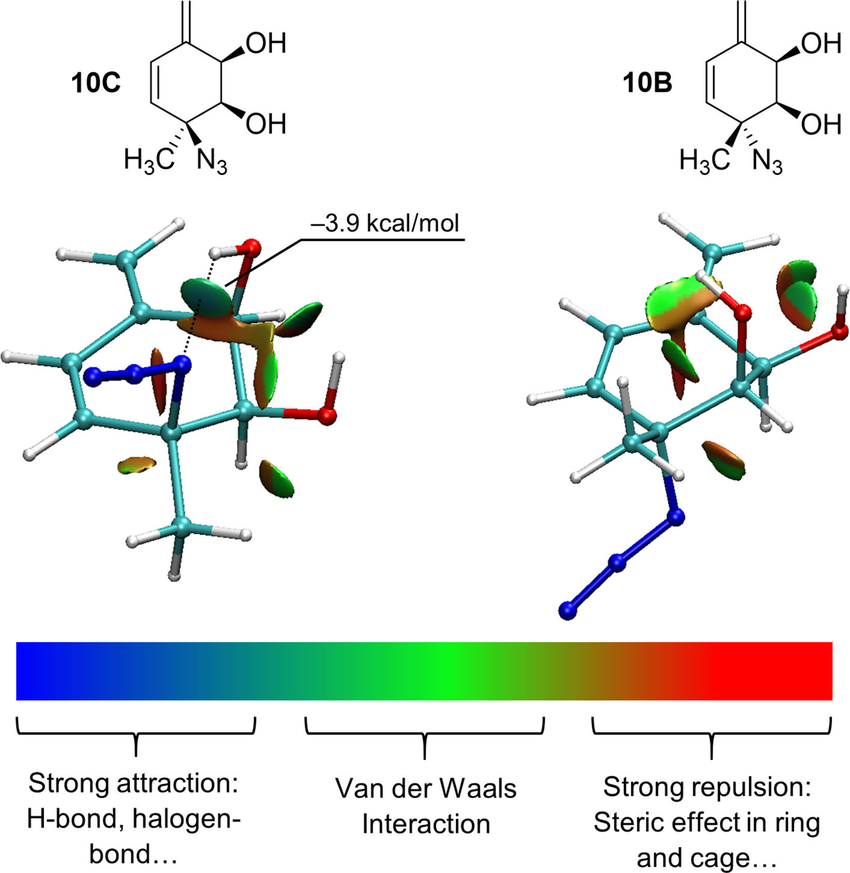
DOI:10.1002/ejoc.202101156
分子动力学与统计模拟的作用
尽管量子化学计算精度高,但其计算成本限制了体系规模与时间尺度。而分子动力学(MD)模拟则为弱相互作用提供了另一种有效途径。
通过构建合适的力场(如AMBER、CHARMM、OPLS),研究者能够在大规模原子体系中追踪弱相互作用随时间的演化。MD不仅能捕捉氢键网络的形成与断裂过程,还能量化疏水相互作用与π–π堆积的统计特征。
在生物体系中,MD结合氢键分析工具(如HBond analysis)、径向分布函数(RDF)与能量分解方法,可以揭示蛋白质–配体识别中的关键弱相互作用位点。此外,粗粒化MD方法则能在更大时间与空间尺度上分析疏水作用与分子自组装机制。
通过这些模拟,弱相互作用不仅被看作一个静态的能量值,更被赋予动态特征,反映其在体系稳定性与功能实现中的复杂作用。
可视化与空间分布分析
除了能量数值,弱相互作用的空间分布特征同样是研究重点。独立梯度模型(IGMH/IGM)与非共价相互作用指数(NCI index)是目前广泛使用的两类方法。
它们通过计算电子密度梯度与简化函数,直观展示弱相互作用区域,并用不同颜色或等值面区分氢键、范德华力与排斥作用。这类方法在图像化展示分子堆积与吸附界面时尤为直观,使研究者能够快速识别关键相互作用位点。
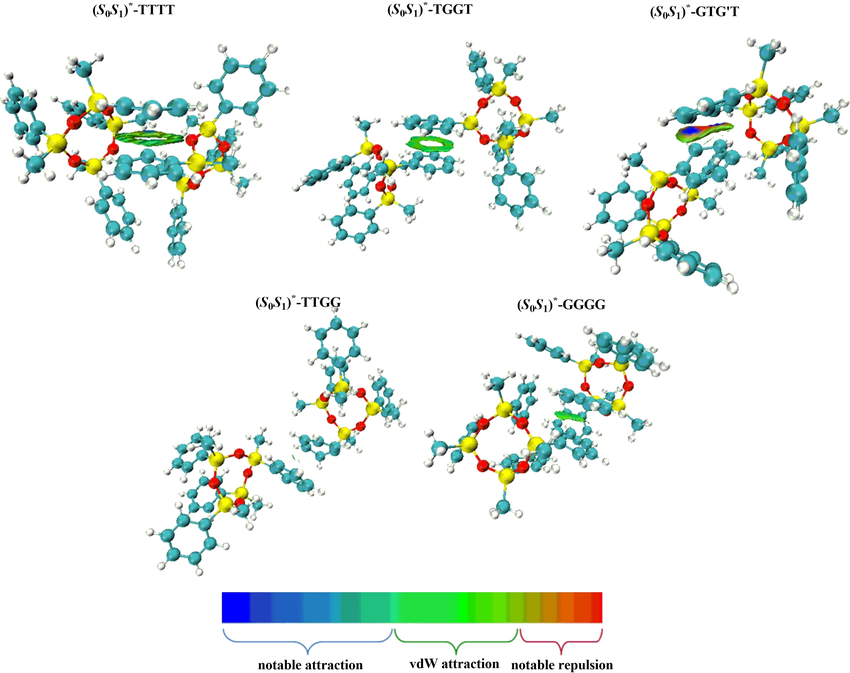
DOI:10.1002/slct.202300097
局域化函数(ELF, Electron Localization Function)与量子理论的原子间相互作用(QTAIM)方法同样可用于弱相互作用分析。
ELF揭示电子对局域化程度,常用于辅助分析氢键特征;QTAIM则通过键临界点(BCP)的电子密度与拉普拉斯值定量刻画弱相互作用的强度与本质。这些空间分布分析方法与能量分解计算结合,能够提供全面而直观的多维度理解。
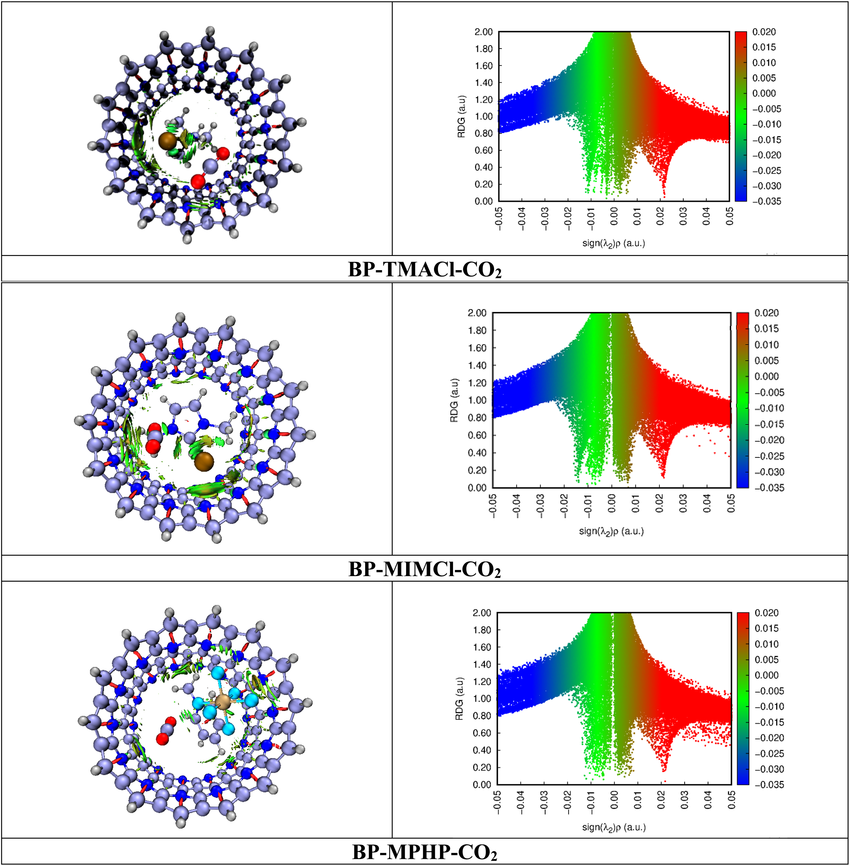
DOI:10.1039/d4ra03394a
实际应用
在生物体系中,弱相互作用决定了蛋白质折叠与配体结合。氢键与疏水作用维持蛋白质三级结构,π–π堆积与偶极作用影响药物分子的结合选择性。
对于材料科学而言,弱相互作用控制了金属有机框架(MOFs)、共价有机框架(COFs)与二维材料中的分子吸附行为,是气体存储、催化反应与分子筛分的关键驱动力。
在表面科学与催化中,弱相互作用常决定吸附构型与能量壁垒。例如,O₂分子在过渡金属表面的弱吸附态为其后续解离提供前驱态;有机分子在二维材料表面的π–π作用调控电荷转移过程。这些应用场景表明,弱相互作用不仅是理论问题,更是理解与设计功能体系的核心工具。
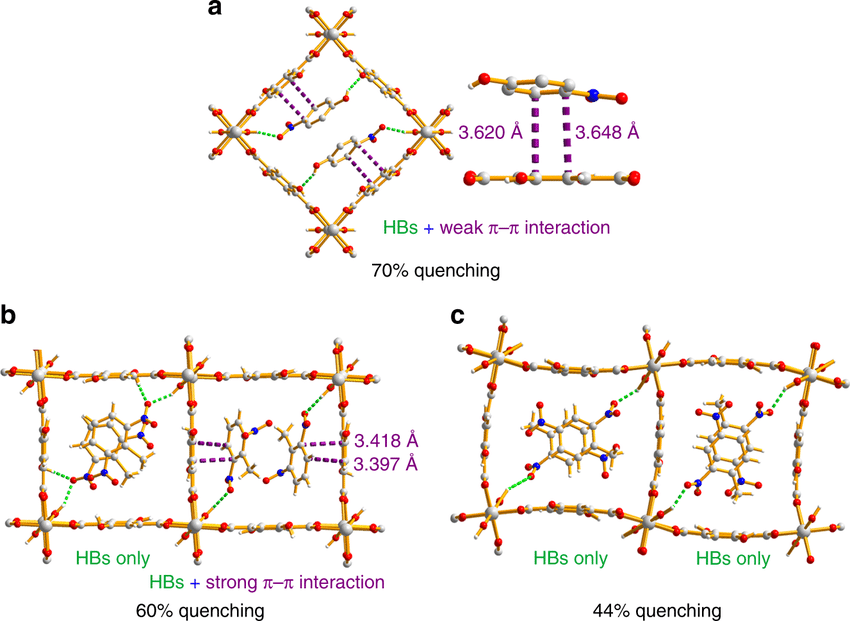
DOI:10.1038/s42004-019-0135-2
未来发展与跨科学意义
随着计算资源与理论模型的发展,弱相互作用分析正在逐渐走向精细化与多尺度化。高精度量子化学方法与机器学习潜力函数的结合,将使得大规模弱相互作用分析成为可能。
此外,实验技术如二维红外光谱、核磁共振(NMR)与扫描隧道显微镜(STM)为理论计算提供验证,使计算与实验的对接更加紧密。未来弱相互作用的研究将更加注重动态性、环境依赖性与协同效应,为多尺度跨学科问题提供解决方案。
在跨学科背景下,弱相互作用研究连接了化学、物理与生物学的多个前沿领域。它既是分子识别与药物设计的基石,又是新材料开发与纳米器件构筑的重要支撑。
随着理论方法不断完善与计算精度不断提高,弱相互作用的研究将不仅停留在解释层面,而是进一步发展为预测与调控复杂体系性质的重要手段。
总结
弱相互作用虽能量较小,却在决定体系结构与功能中扮演不可替代的角色。通过量子化学方法、分子动力学模拟、能量分解分析与空间分布可视化,研究者能够从多个维度揭示其本质与机制。
弱相互作用研究不仅深化了我们对分子间与材料界面行为的理解,也为药物设计、材料工程、催化机制等应用提供理论指导。
未来,随着跨学科方法的融合与数据驱动模型的发展,弱相互作用的研究将持续拓展,并在科学与技术的多个领域发挥更加广泛而深远的影响。