

说明:本文华算科技将从定义出发,系统介绍计算方法、应用实践、关键点,从而帮助读者全面掌握弱相互作用的解析框架。
弱相互作用(Weak Interactions)是分子科学与材料物理中的核心课题,与强相互作用(如共价键或离子键)不同,弱相互作用源于电子云分布、静电效应和量子色散的综合作用,具有高度动态性和协同性。
近年来,随着计算化学的发展,高精度理论工具如密度泛函理论(DFT)、分子动力学(MD)和可视化模型(如RDG、IGMH)已能深入解析这些作用机制。


什么是弱相互作用?
弱相互作用是指分子间或分子内非共价键的吸引力,主要包括氢键(如O-H···O)、范德华力(van der Waals forces)、π-π堆积(如苯环间的堆叠)、疏水作用和偶极–偶极相互作用等。
图1分子间弱相互作用的分类 DOI:10.1021/acsami.3c04582
这些作用的能量虽仅为共价键的1/10至1/100(约1–10 kcal/mol),但通过协同效应,能在多体体系中产生显著影响。
图2氢键网络与π-π堆积(隐含于范德华力中)共同维持DNA稳定性的机制
DOI:10.1021/acs.jpclett.5b01899
例如,在DNA双螺旋中,氢键网络和π-π堆积共同维持结构稳定性;在金属有机框架(MOF)材料中,范德华力调控孔隙率和吸附性能。物理本质上,弱相互作用源于电子密度离域和瞬时偶极矩,其强度受环境因素(如溶剂极性)强烈影响。
从微观视角看,氢键涉及氢原子与高电负性原子(如O、N)的部分静电吸引,范德华力包括色散(瞬时偶极诱导)和诱导作用,而π-π堆积则依赖芳香环的电子云重叠。
这些类型在生物体系(如蛋白质折叠)和材料科学(如催化剂设计)中无处不在。例如,药物分子与靶标蛋白的结合常依赖多重弱相互作用的协同,单个作用能虽低,但集体贡献可达几十kJ/mol,决定结合亲和力。理解这些定义是解析弱相互作用的基础,需结合量子力学和统计物理视角。
弱相互作用的重要性体现在其跨学科价值:在自然界中,它驱动分子自组装和生物进化;在科技领域,它支撑药物设计、纳米材料开发和能源存储。
实验上直接测量困难(因能量微弱),但理论计算可量化其贡献。后续章节将深入计算方法,揭示如何从电子密度层面捕捉这些“看不见的力”。
弱相互作用的计算分析方法
计算弱相互作用的核心在于高精度理论工具,可分为量子化学方法、分子动力学模拟和可视化技术三大类。
量子化学方法提供电子层面解析,例如密度泛函理论(DFT)常用于基础计算,但对色散作用(范德华力)常低估,因此发展了DFT-D2/D3修正和vdW-DF函数,引入色散效应。对称适应微扰理论(SAPT)则能将总相互作用能分解为静电、交换、诱导和色散分量,量化各贡献比例。
例如,在药物–受体体系中,SAPT可区分氢键和疏水作用的能量占比,精度达0.1kcal/mol。这些方法依赖量子化学软件如Gaussian或ORCA,计算成本较高,适用于中小分子体系。
RDG方法
约化密度梯度(RDG)分析是一种可视化工具,通过归一化电子密度梯度(RDG ≈0的区域对应作用位点)实现弱相互作用空间定位。结合sign(λ₂)ρ参数,蓝色凹陷表示强吸引(如氢键),绿色区域表征范德华力。
电子密度分析(ρ(r))则量化键临界点(BCP)的ρ值和拉普拉斯量(∇²ρ):氢键的ρ(BCP)通常为0.002–0.035a.u.,且∇2ρ>0(电子离域),而共价键∇2ρ(电子集中)。
自然键轨道(NBO)分析通过二阶微扰能E(2)评估超共轭效应,如供体孤对电子与受体σ*轨道的相互作用,E(2)值约5kcal/mol时可标识关键电荷转移路径。
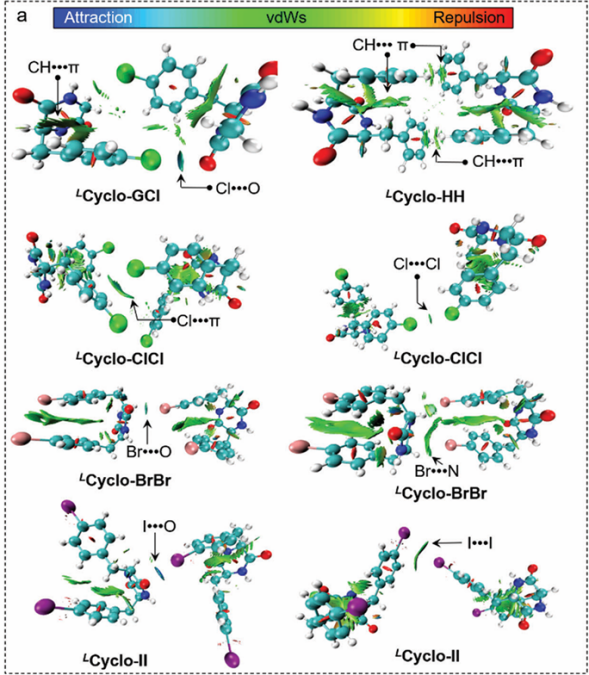
图3可视化和区分分子间和分子内发生的NCI和RDG图
DOI: 10.1002/smll.202302517
分子动力学方法
分子动力学(MD)模拟扩展了时间尺度,使用力场如AMBER或CHARMM追踪弱相互作用的动态演化。MD可量化氢键网络的形成/断裂,通过径向分布函数(RDF)统计疏水作用或π-π堆积的概率。例如,在蛋白质折叠模拟中,MD结合氢键分析工具可识别稳定构象的关键位点。
IGMH和NCl方法
可视化方法如独立梯度模型(IGMH)和非共价相互作用指数(NCI)通过电子密度梯度生成等值面图,用颜色区分作用类型(红色为排斥,蓝色为吸引)。
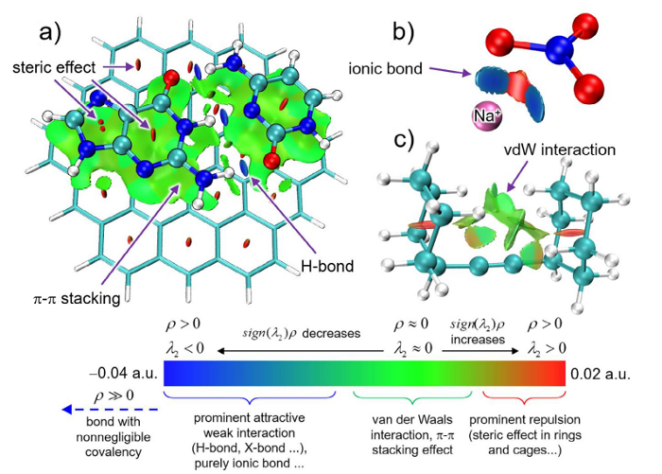
图4突出非共价相互作用的代表性系统的NCI图
DOI: 10.1002/anie.202504895
相互作用区域指示符(IRI)则无需预设类型,直接显示共价键和弱相互作用区域,灵敏度达0.5kcal/mol。Hirshfeld表面分析基于原子范德华半径,通过dnorm参数(负值表示强接触)和形状指数(凸–凹模式指示π-π堆积)提供晶体学视角。
计算流程通常分步:先用量子化学优化几何结构,再用RDG或IGMH可视化作用区域,最后通过MD或SAPT进行动态或能量分解。
工具选择取决于体系规模——小分子用SAPT追求精度,大生物分子用MD平衡效率。计算中的关键挑战是处理环境效应(如溶剂化)和协同性,需结合机器学习潜力函数提升效率。
图5 IRI分析弱相互作用图 DOI: 10.1002/anie.202504895
弱相互作用的应用
弱相互作用的解析在药物设计中至关重要,通过量化氢键、π-π堆积和疏水作用,优化药物–靶标结合亲和力。
例如,在抗病毒药物研发中,NBO分析和MD模拟可识别关键残基的E(2)能值,预测结合自由能;
RDG可视化则帮助设计分子结构以增强协同效应。临床案例显示,HIV蛋白酶抑制剂的设计依赖多重弱相互作用网络,将结合能提升20–30%,减少副作用。应用流程包括:计算蛋白–配体界面的弱作用能,筛选候选分子,再通过实验验证。
在材料科学领域,弱相互作用驱动金属有机框架(MOF)和共价有机框架(COF)的孔隙调控与气体吸附。例如,MOF材料中,范德华力和π-π堆积主导甲烷存储能力;
IRI分析可量化孔隙表面作用能,关联到吸附等温线。实际应用中,Hirshfeld表面统计接触占比(如氢键占表面积百分比)指导材料合成,提升CO2捕获效率30–50%。此外,二维材料(如石墨烯)的表面催化依赖弱吸附态,DFT计算揭示O2分子的范德华作用如何降低反应能垒,应用于燃料电池
设计。
生物体系中,弱相互作用维持蛋白质三级结构和DNA稳定性。氢键网络和疏水作用确保蛋白质折叠正确,MD模拟可追踪动态过程;在DNA复制中,π-π堆积和氢键协同作用通过NBO分析验证。
应用扩展到疾病机制研究,如阿尔茨海默症中淀粉样蛋白聚集的分子动力学模拟。未来,弱相互作用解析将整合人工智能,预测复杂体系行为,推动个性化医疗和绿色材料创新。
弱相互作用解析的核心要素
协同效应是弱相互作用的核心特征,单个作用能虽弱,但集体贡献可主导体系行为。例如,在DNA或MOF中,多重氢键和π-π堆积的叠加能将稳定能提高数倍。计算中必须通过SAPT或EDA分解能量分量,避免低估;
实验上,核磁共振(NMR)或STM可验证协同性。关键点在于模型选择——简单体系用DFT-D3,复杂体系用MD,确保精度与效率平衡。
计算精度依赖于方法校正和环境建模。色散作用易被标准DFT忽略,需引入MBD修正;溶剂效应通过隐式或显式模型处理,如PCM溶剂化。
可视化工具如RDG或IGMH是诊断关键,但需结合QTAIM(量子理论原子间作用)分析BCP点,量化ρ(BCP)和∇2ρ值。例如,氢键的ρ(BCP)>0.02a.u.时强度显著。实践中,多尺度方法(量子化学/MD耦合)和机器学习加速计算成为趋势,提升大体系处理能力。
实验与理论结合是验证核心。二维红外光谱或X射线衍射提供结构数据,与计算预测对比;未来方向包括动态原位表征和高通量筛选。最终,弱相互作用研究需跨学科整合,从量子物理到生物材料,推动基础发现向应用转化。
总结
弱相互作用解析是计算化学的支柱,从定义到应用,揭示了微观世界的“看不见的力”。通过量子化学、分子动力学和可视化工具,我们能量化氢键、范德华力等作用,驱动药物设计、材料创新和生物研究。
关键点如协同效应和精度控制强调方法选择的重要性;顶刊案例证明理论指导实践的威力。未来,随着AI和多尺度模拟发展,弱相互作用研究将更精细化、动态化,赋能碳中和、精准医疗。