

弱相互作用(noncovalent interactions, NCI)是指不伴随传统共价键形成、能量相对较低但在化学与材料体系中广泛存在的分子间或分子内作用力。
其能量范围通常为数 kJ/mol 至数十 kJ/mol,虽单个作用较弱,但在生物大分子折叠、分子自组装、晶体工程、表界面稳定以及催化反应选择性等方面具有决定性作用。
常见的弱相互作用类型包括氢键、范德华力/色散作用、π–π 堆积、C–H···π 相互作用、卤键、阳离子–π 作用等,不同类型在物理本质上可归因于静电、感应、色散及交换排斥等相互竞争与协同的贡献。
弱相互作用
在化学体系中,原子和分子之间的相互作用可以分为两大类:强相互作用(如共价键、离子键、金属键)和弱相互作用(noncovalent interactions, NCI)。
弱相互作用的特点是能量较低(通常几个 kJ/mol 到几十 kJ/mol),方向性可强可弱,而且作用范围相对较长(尤其是静电型相互作用)。虽然每一对作用的能量不大,但在多分子体系或大分子体系中,大量弱相互作用的协同效应可以产生决定性的稳定化和构象控制作用。
在计算化学研究中,弱相互作用的分析有三个重要意义:
解释结构稳定性:比如为什么某个晶体结构稳定、某种分子构象更有利,往往是弱相互作用网络决定的。
揭示反应选择性:催化反应的过渡态中,弱相互作用能降低特定构象的能垒,从而提高区域或立体选择性。
关联宏观性质:材料的层间结合能、蛋白质折叠、分子自组装等宏观现象,都和微观弱相互作用直接相关。
因此,在计算化学中,“弱相互作用”不仅是一个物理化学概念,还涉及到多种计算与分析工具,例如能量分解分析(SAPT, EDA)、拓扑分析(QTAIM)、非共价相互作用指数(NCI)、独立梯度模型(IGM/IGMH)、静电势映射(ESP)、差分电荷密度(Δρ)等。
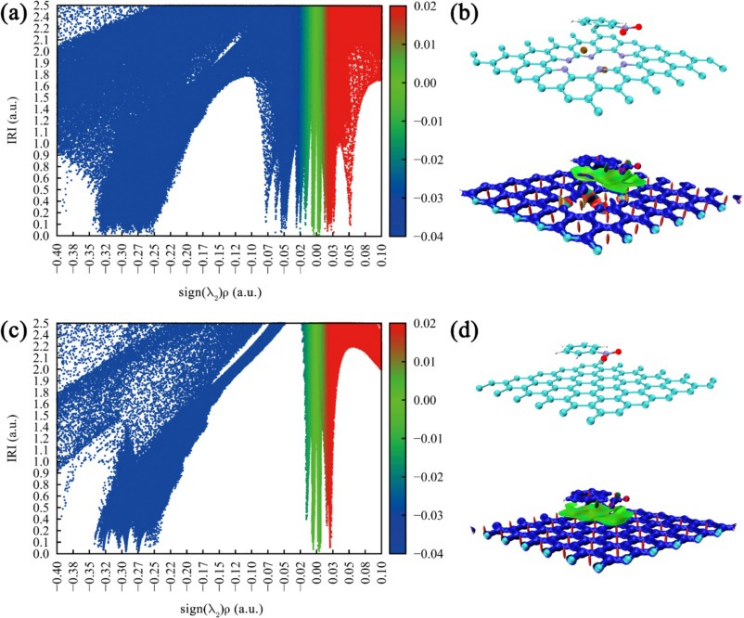

DOI:10.1016/j.chemphys.2024.112513
弱相互作用的分类
弱相互作用的分类方法有多种,常见的分法是按照物理本质或几何特征来划分。下面按计算化学和化学直觉相结合的方式,把主要的弱相互作用类型展开说明。
静电相互作用(Electrostatic interactions)
静电相互作用源于分子间或分子内部不同电荷分布之间的库仑作用,包括点电荷之间、偶极-偶极、偶极-四极、四极-四极等多极相互作用。它是很多弱相互作用的主导稳定化来源,尤其是在极性体系中。
在能量分解分析(SAPT、EDA)中,静电项通常是通过片段的静态电荷密度计算得到的,与电子极化无关。它的强度依赖于电荷分布、偶极矩大小以及相对取向。
典型实例
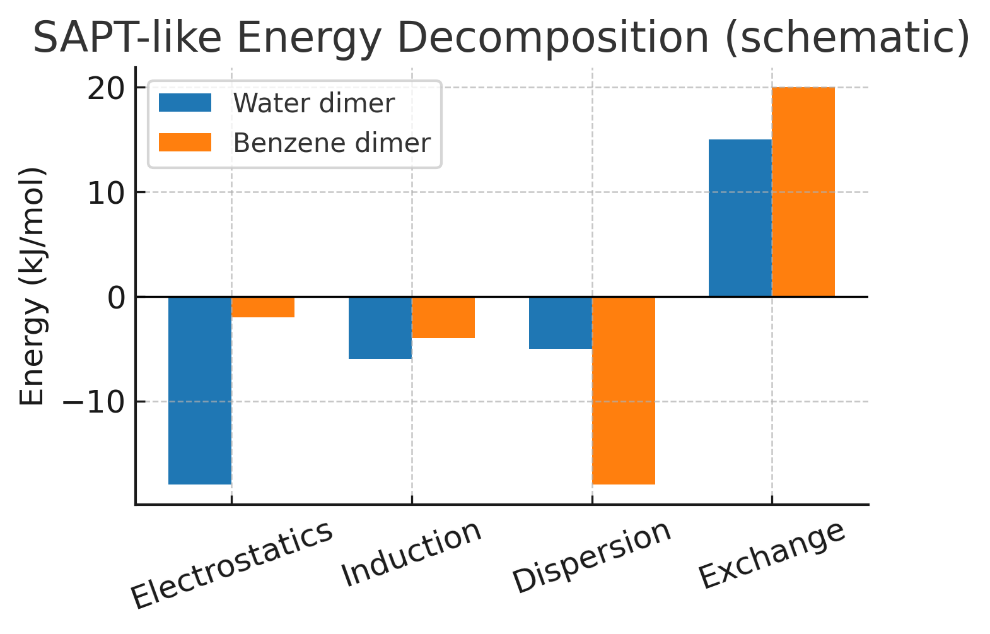
水二聚体中的氢键,静电作用占主要稳定化贡献。
蛋白质-配体结合中,带电氨基酸侧链与配体极性基团的长程吸引。
氢键(Hydrogen bond)
氢键是一种特殊的静电-极化耦合型相互作用,通常记作 D–H···A(供体–氢–受体)。供体 D 上的氢原子与受体 A(常含孤对电子)形成取向性较强的相互作用。
氢键在 QTAIM 分析中常对应于供体氢和受体原子之间的一个键鞍点(BCP),电子密度 ρ(BCP) 约 0.002–0.035 a.u.。能量分解中,静电和感应贡献显著,色散较小但不可忽视。
分类
传统氢键:如 O–H···O、N–H···O,键能通常 10–40 kJ/mol。
弱氢键/非传统氢键:如 C–H···O、C–H···π,键能通常 1–5 kJ/mol,但在大分子折叠中可累积产生显著影响。
双氢键、协同氢键:多个氢键同时作用产生协同效应。
范德华力(van der Waals forces)与色散相互作用(Dispersion)
范德华力包括诱导偶极-诱导偶极(色散)、偶极-诱导偶极(感应)以及取向作用等。狭义上,色散作用是由瞬时偶极及其相关性引起的量子力学效应。
物理本质:色散能量在长程衰减为−C6/R6,C6系数与分子极化率有关。它在非极性体系中是主要吸引力,在极性体系中则是静电作用的补充。
计算化学上高精度从头算(CCSD(T))或SAPT可以直接给出色散分量。DFT需要通过经验修正(如 DFT-D3/D4、VV10)或非局域泛函引入色散效应。
实例
苯二聚体的π–π 堆积:色散是主要稳定化来源。
惰性气体分子之间的结合。

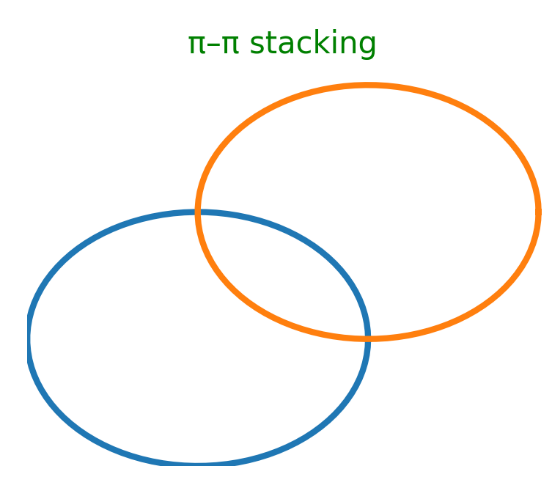
π–π 相互作用(π–π stacking)
π–π 相互作用是芳香体系之间的弱吸引,包括平行堆积(sandwich)、平行位移(slipped-parallel)和 T 形等构型。
物理本质:主要由色散作用稳定,也包含静电(四极矩)和少量感应成分。不同构型的能量差异主要来自交换排斥与静电分布差异。
计算化学上EDA/SAPT 可量化各构型的色散/静电比例。NCI 图像中,π–π 区域常呈现大面积绿色等值面(范德华型)。
应用
核酸碱基对堆积稳定性。
有机半导体分子在固态的堆积模式调控。
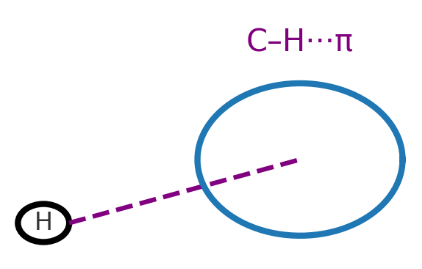
C–H···π 相互作用
供体是 C–H 基团,受体是芳香 π 体系。虽然单个作用能弱(1–3 kJ/mol),但在大分子和晶体中数量多时可显著影响构象。
物理本质:主要是 C–H 偏正与 π 面电子云的静电吸引,以及少量色散作用。
卤键(Halogen bond)
卤原子(Cl、Br、I 等)在共价键延伸方向具有电子密度缺陷(σ-hole),可与富电子中心(如 O、N、π 云)发生定向吸引。
物理本质:静电作用为主(σ-hole 区正电势),感应与色散为辅。
硫键、硫–π、硫–卤相互作用
S 原子在共价键延伸方向也可能有 σ-hole,或通过孤对与 π/卤素形成吸引作用。在含硫药物、蛋白质中常见,对构象稳定与活性位点几何有影响。结合 NBO(自然键轨道分析)、EDA、QTAIM 等,可区分金属–配体间的共价成分与弱相互作用成分。
疏水作用(Hydrophobic effect)
疏水作用本质上并不是单一的微观力,而是水环境中非极性分子/基团倾向聚集以减少界面面积的现象。它涉及水分子的氢键网络重排和熵效应。计算化学难点疏水效应更多体现为溶液热力学整体的自由能变化,需要分子动力学(MD)结合自由能计算(FEP、TI、MM-PBSA 等)分析。
其他类型
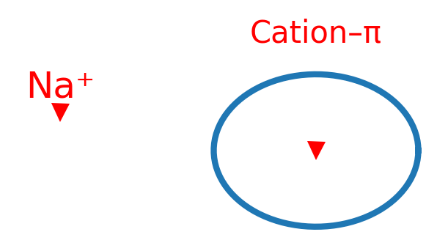
阳离子–π 相互作用:阳离子与芳香π 面之间的静电–色散协同吸引。
阴离子–π 相互作用:依赖π 面的正静电势区,常在电子缺乏的芳环中出现。
多极相互作用:四极–四极、偶极–四极等。
计算化学中的识别与量化
不同类型的弱相互作用往往在以下分析中有特征信号:
能量分解分析(EDA、SAPT)
把总相互作用能拆成静电、感应、色散、交换等分量,直观看到哪种成分占主导。
QTAIM 拓扑分析
判断是否存在 BCP,测量 ρ、∇2ρnabla^2rho、能量密度 H 等,区分闭壳层(弱相互作用)与共价型相互作用。
NCI、IGM/IGMH 可视化
用电子密度和梯度信息在三维空间中画出相互作用区域的等值面,并根据 sign(λ2)ρ颜色区分吸引/排斥。
静电势映射(ESP)
在分子表面映射静电势,找到σ-hole、π-hole 等热点,预测可能的相互作用位点。
差分电荷密度(Δρ)
对比复合物与各片段的电子密度差,直观显示电子转移方向和空间分布。
能量尺度参考
强氢键(O–H···O):20–40 kJ/mol
中等氢键(N–H···O):10–30 kJ/mol
弱氢键(C–H···O、C–H···π):1–5 kJ/mol
π–π 堆积:2–10 kJ/mol
卤键:5–20 kJ/mol
阳离子–π:10–80 kJ/mol(取决于离子和 π 面性质)
纯色散(如稀有气体二聚体):
总结
弱相互作用虽然单个能量小,但在生物大分子稳定性、晶体结构、自组装、材料层间作用、催化反应选择性等方面,往往起决定性作用。
它们包括静电相互作用、氢键、范德华/色散、π–π 堆积、C–H···π、卤键、硫键、阳离子–π、阴离子–π、疏水作用等多种类型,每种都有其物理本质和特征参数。
在计算化学中,通过能量分解、拓扑分析、非共价相互作用可视化、静电势映射、差分电荷密度等方法,可以识别、分类和定量这些作用,为理解分子行为和设计新材料/药物提供了坚实的理论支持。