

说明:静电势是描述单位正电荷势能的标量场,源于原子核与电子云的叠加贡献,具有保守场、可叠加性等特性,是连接微观电子结构与宏观相互作用的核心量。
其计算方法涵盖DFT、PB方程、多极子展开等,前沿借助机器学习、非局部介电理论及GPU加速提升效率与精度,应用于生物分子、材料界面等领域,经典案例揭示微管静电势与细胞运输机制的关联。
什么是静电势?
静电势(Electrostatic Potential, ESP)是描述静电场中单位正电荷所具有势能的标量场,其核心定义可通过电场强度的线积分表达。
在量子化学与分子模拟的框架下,静电势的物理本质源于原子核与电子云的共同贡献,清晰揭示了静电势的双重起源——带正电的原子核产生吸引势能,而带负电的电子云则产生排斥势能,两者的叠加决定了空间某点的静电势数值。
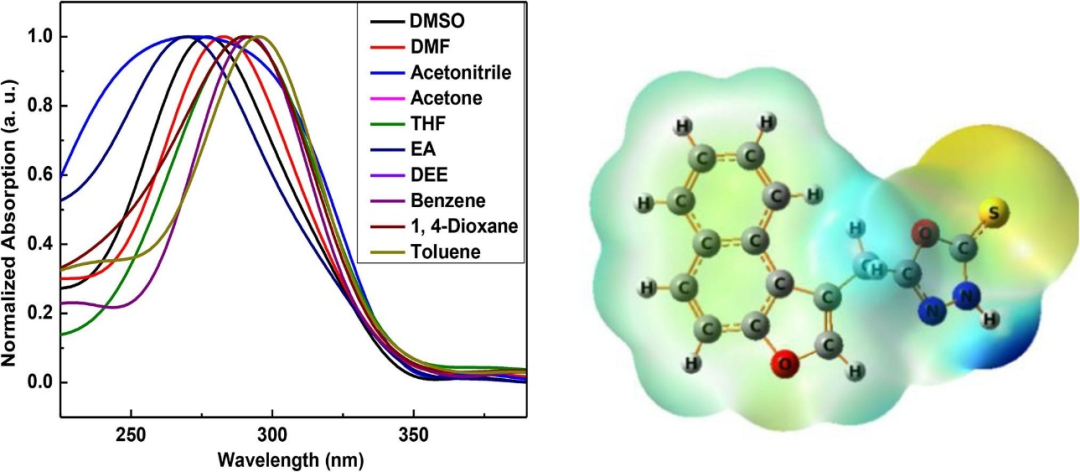

DOI:10.1016/j.rechem.2023.101046
静电势具有三个关键特性:其一,作为保守场,它满足泊松方程∇²φ=-ρ/ε₀,其中ρ为总电荷密度,ε₀为真空介电常数,这一特性确保了静电势的分布可通过电荷密度唯一确定,为理论计算提供了数学基础。
其二,可叠加性,多粒子系统的静电势等于各组分单独存在时产生的静电势的线性叠加,即φ_total (r)=Σφ_i (r),这使得复杂体系的静电势计算可分解为对单个原子或分子的计算,大幅简化了问题复杂度。
其三,明确的物理意义,φ(r)的数值直接反映空间某点对带正电探针的吸引或排斥趋势——负值区域因电子云密度较高,对正电荷探针表现出较强吸引力,易与阳离子发生相互作用;正值区域则因核电荷暴露或电子云稀疏,对正电荷探针产生排斥,更易吸引阴离子。
例如,在蛋白质分子中,氨基酸残基的侧链往往形成局部正负电势区域:赖氨酸的氨基(-NH₃⁺)周围呈现正电势,易与带负电的磷酸基团结合;天冬氨酸的羧基(-COO⁻)周围则为负电势,可与金属阳离子配位。
这些特性使得静电势成为连接微观电子结构与宏观分子间相互作用的核心物理量,在理解分子识别、催化反应、材料界面行为等领域具有不可替代的作用。
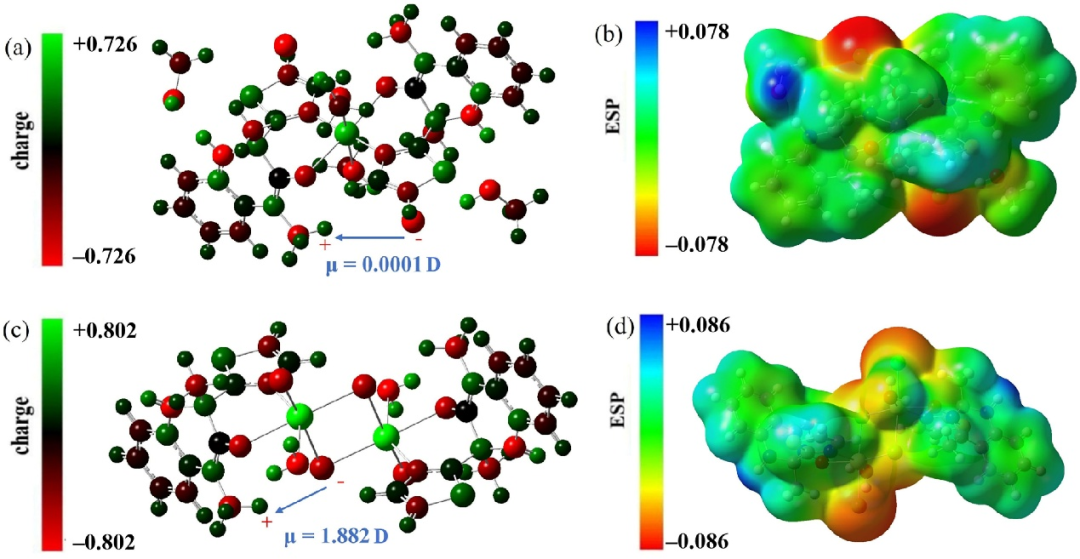
DOI:10.1016/j.molstruc.2025.143034
怎么去算静电势?
静电势的计算方法随研究体系的尺度与复杂度发展出多套理论框架,涵盖量子化学精确计算、连续介质模型及简化近似方法,各自在精度与效率间取得平衡,以适应从原子级电子结构到生物大分子体系的广泛需求。
在量子化学框架下,密度泛函理论(DFT)是计算静电势的主流方法,其核心流程为通过求解Kohn-Sham方程获得体系的电子密度ρ(r),再基于ρ(r)计算静电势。
具体而言,静电势可分解为核贡献项与电子密度泛函项:φ(r)=Σ(Zₐ/|r-Rₐ|)+φₑₗₑc (r),其中φₑₗₑc (r)=-∫ρ(r’)/|r-r’|dr’为电子云产生的静电势。
为提升计算精度,广义梯度近似(GGA)泛函通过引入电子密度的梯度修正,比局域密度近似更准确地描述电子云的非均匀分布,尤其在分子界面或缺陷附近。
投影缀加波(PAW)方法则通过赝势与全电子波函数的转换实现高效计算,其核心思想是将电子波函数分为芯区与价区,芯区采用赝势描述,价区则保留全电子特性,表达式为ψₐ(r)=φₐ(r)+Σp (θₐ(r)(χₚ(r)-φₚ(r))),其中φₐ为赝波函数,χₚ为全电子波函数,θₐ为球谐截断函数。
这种方法在保留核心电子对静电势贡献的同时,大幅降低计算成本,适用于包含重元素的复杂体系。
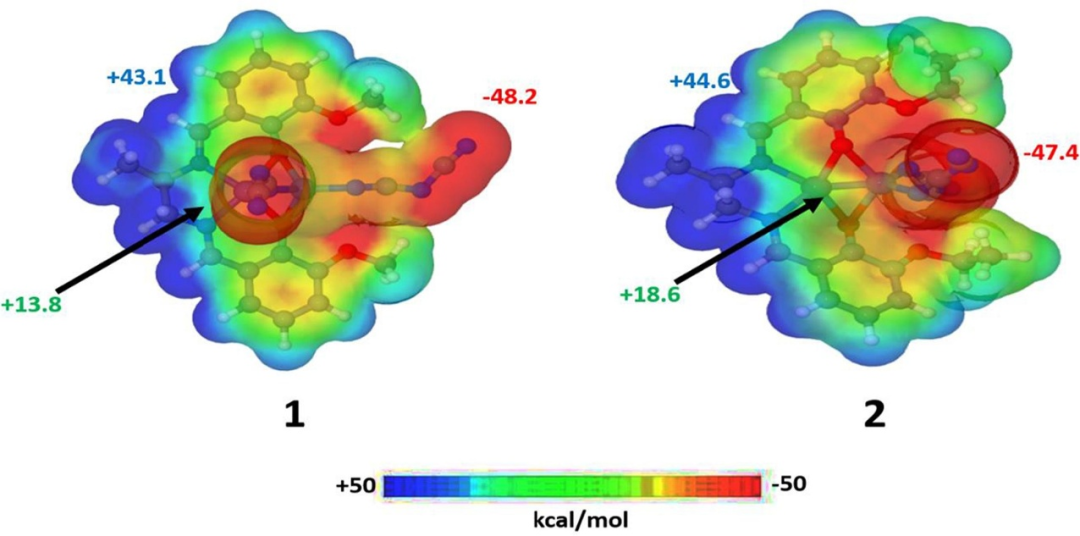
DOI:10.1016/j.ica.2019.02.022
针对生物大分子的溶液体系,连续介质模型中的Poisson-Boltzmann(PB)方程成为首选工具,其方程形式为∇・(ε(r)∇φ(r))-κ²(r) sinh (φ(r))=-ρ_fixed (r)/ε₀,其中ε(r)为位置依赖的介电常数,κ(r)为Debye-Hückel参数,ρ_fixed (r)为固定电荷密度,sinh项描述离子的Boltzmann分布。
为提升求解效率,算法层面发展出自适应笛卡尔网格法,通过在分子界面处加密网格,在保证精度的同时减少计算量;快速多极子法(FMM)则通过将远场相互作用近似为多极子展开,将计算复杂度从O(N²)降至O(N),使核糖体等超大体系的静电势计算成为可能。
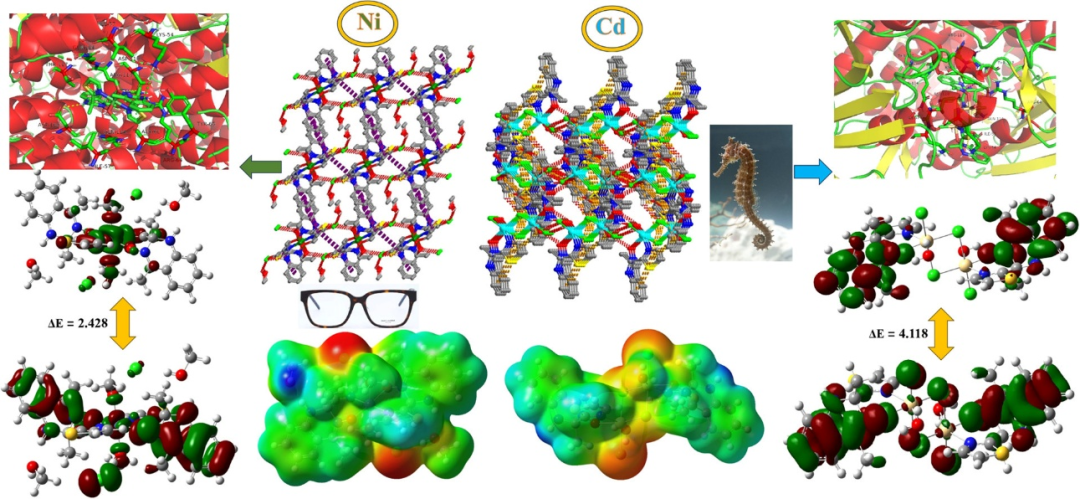
DOI:10.1016/j.molstruc.2025.143034
对于追求极致效率的高通量筛选,多极子展开与可转移原子模型提供了简化方案,其核心是将原子的静电势贡献近似为多极矩的叠加:φ(r)=Σ(qₐ/|r-Rₐ|+μₐ・(r-Rₐ)/|r-Rₐ|³+… ),其中qₐ、μₐ分别为原子的电荷、偶极矩。
TAAM(Transferable Aspherical Atom Model)数据库通过预存不同化学环境下原子的多极参数,可快速重建蛋白质等生物分子的静电势,计算速度比DFT快1-2个数量级,虽精度略有损失,但能满足大规模筛选需求。
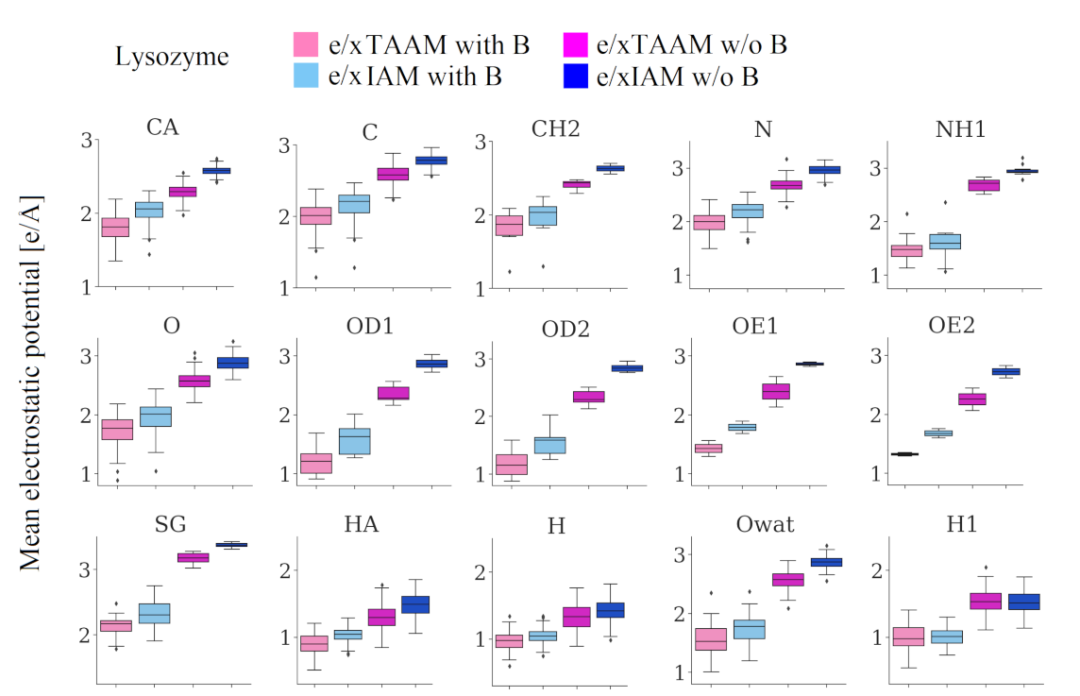
DOI:10.1107/S2059798322005836
这些方法的协同应用,使得静电势计算能够覆盖从原子级电子结构到宏观生物体系的全尺度研究,为不同领域的理论分析提供灵活工具。
静电势前沿
静电势计算的前沿研究正通过机器学习、理论模型创新与计算技术升级,突破传统方法的精度与效率瓶颈,拓展其在复杂动态体系中的应用边界。
机器学习驱动的静电势预测是当前最具活力的方向之一,图神经网络(GNN)如SchNet模型通过学习分子的拓扑结构与静电势场的映射关系,可直接从分子结构预测三维空间中的静电势分布。
其核心优势在于摆脱了传统量子化学计算对电子密度的依赖,通过训练集优化网络参数,预测速度较DFT计算提升100倍以上,且误差可控制在5%以内。这种方法特别适用于高通量筛选,例如在药物分子设计中,可快速评估候选分子与靶蛋白的静电相互作用能,加速先导化合物的发现。
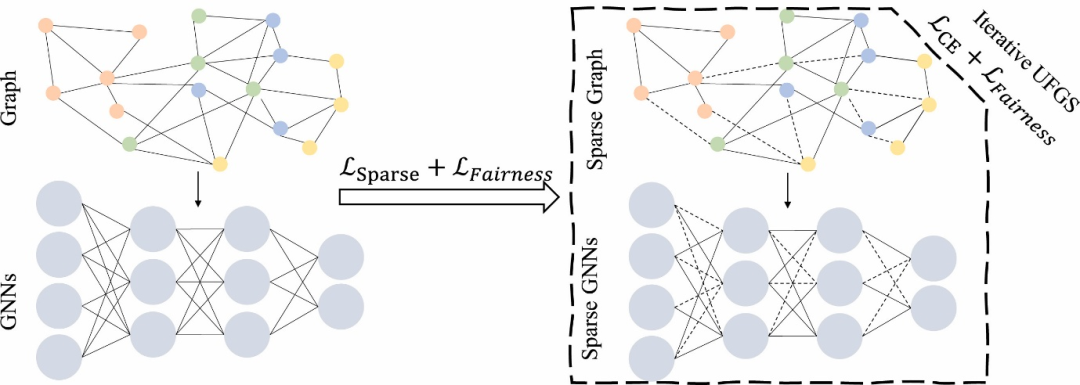
DOI:10.1016/j.neucom.2025.130641
非局部介电响应理论则针对经典连续介质模型的局限性进行修正,传统PB方程假设介电响应是局部的,但在分子界面,氢键网络的协同作用会导致介电响应具有空间色散性。
新理论引入关联长度ξ描述这种非局部效应,这一修正显著改善了界面处静电势的计算精度,例如在DNA双链表面,非局部理论计算的静电势与实验测量的X射线散射数据吻合度比传统PB方程提高20%,更准确反映了溶剂化效应对静电势的调制。
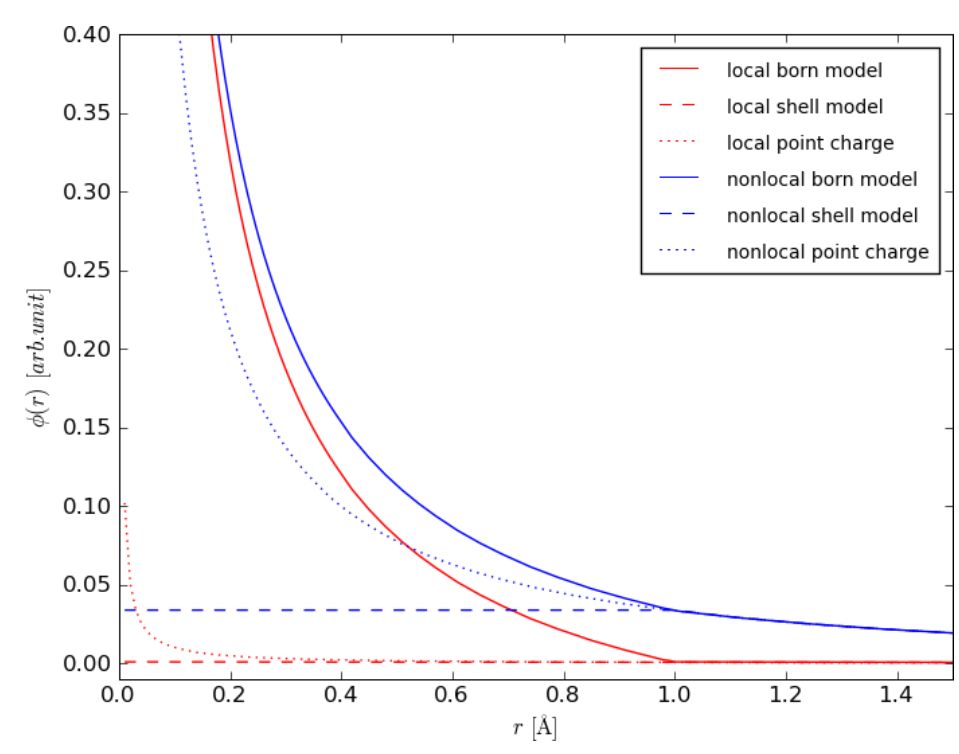
GPU并行化计算技术则为动态体系的静电势追踪提供了硬件支撑,如DelPhiForce工具链通过多尺度近似与GPU的并行计算能力,实现微秒级分子动力学模拟中静电势的实时更新。
其核心机制是利用GPU的thousands of cores同时处理网格点的电势计算,将每步分子动力学的静电势更新时间从CPU的秒级缩短至毫秒级,使得研究蛋白质构象变化过程中静电势的动态演化成为可能。
例如,在肌球蛋白与肌动蛋白的相互作用模拟中,GPU加速的静电势计算揭示了构象变化如何通过改变表面电势分布调节结合强度,为肌肉收缩机制提供了新见解。
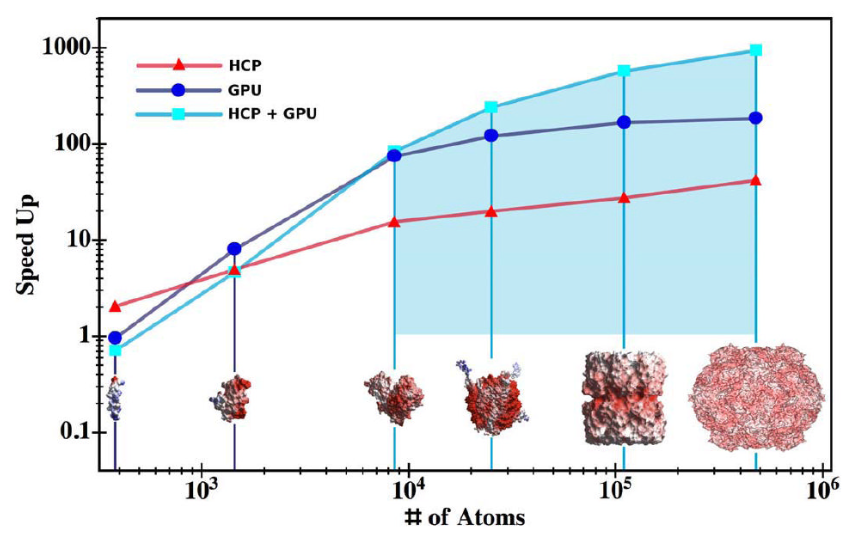
DOI:10.1016/j.jmgm.2010.04.001
这些前沿进展不仅提升了静电势计算的性能,更拓展了其应用场景,使其从静态结构分析迈向动态功能解析,为复杂体系的理论研究提供了更强有力的工具。
微管静电势应用
以文献《Electrostatics of nanosystems: Application to microtubules》为例,该研究通过系统的静电势计算,首次在原子尺度揭示了微管表面的静电势分布规律,阐明了其与带正电运动蛋白的相互作用机制,为理解细胞内物质运输的分子基础提供了关键理论依据。
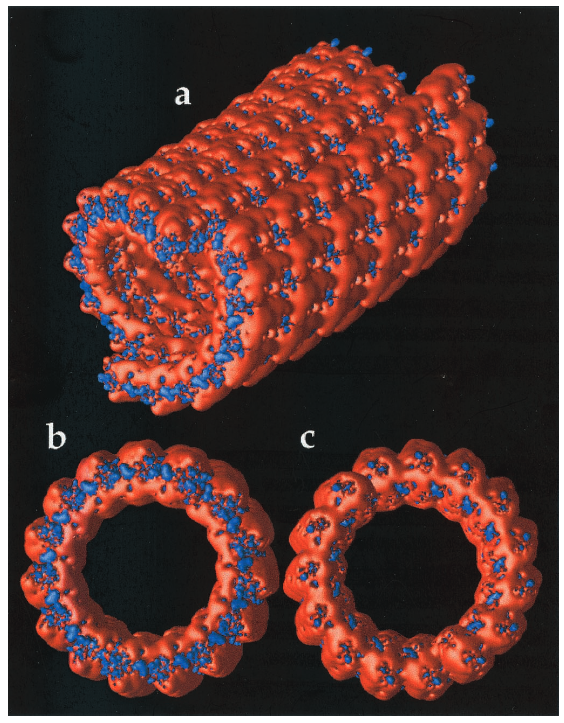
DOI:10.1073/pnas.181342398
计算目标聚焦于微管这一细胞骨架的核心组件,其由α/β微管蛋白异二聚体纵向排列形成中空管状结构,直径约25 nm,长度可达微米级,是驱动蛋白沿其移动并运输囊泡的轨道,研究旨在通过静电势分布解释驱动蛋白的定向运动机制。
方法流程严谨且层次分明:首先进行结构准备,从PDB数据库获取微管蛋白的晶体结构(PDB ID: 1TUB),包含13个异二聚体组成的原丝,通过量子化学优化确定氢原子的精确位置;随后进行电荷分配,采用PDB2PQR工具将蛋白质结构转换为带电荷的模型,分配AMBER力场中的原子电荷。
接着求解PB方程,使用APBS软件进行数值计算,网格参数设置为0.5 Å分辨率以确保界面处的计算精度,蛋白内部的介电常数设为2,溶剂区(水相)设为80,离子强度为150 mM,采用自适应有限差分法处理不规则的分子边界;最后通过可视化软件展示静电势等值面,其中蓝色代表负电势区域,红色代表正电势区域。
关键图表解析揭示了静电势分布的三个核心特征及其生物学意义:微管外表面的深蓝区域对应强负电势,主要由β微管蛋白上的天冬氨酸、谷氨酸残基贡献,这种负电势可通过静电吸引与驱动蛋白的阳离子结构域结合,为驱动蛋白提供初始锚定位点,并指导其沿微管的运动方向。
微管内腔的红色区域呈现正电势,源于α微管蛋白内部的精氨酸残基,可排斥胞质中的阳离子(如Na⁺、K⁺),维持管腔内部的低离子浓度,为微管的结构稳定性提供环境支撑;纵向条纹的交替分布与微管蛋白异二聚体的排列周期一致,这种周期性电势场与驱动蛋白的步进机制相匹配,通过静电势能的周期性变化为驱动蛋白的定向运动提供能量梯度,确保其沿微管长轴单向移动而不反向滑动。
该研究的理论价值在于首次建立了微管静电势的纳米级周期性与细胞内运输功能的直接关联,不仅解释了驱动蛋白运动的定向性与高效性,更为后续抗有丝分裂药物的设计提供了靶点——通过修饰微管表面的负电势区域,可干扰驱动蛋白的结合,抑制细胞分裂,为癌症治疗提供新策略。
其方法论也成为生物大分子静电势研究的典范,推动了静电势计算在细胞生物学领域的广泛应用。
DOI:10.1073/pnas.181342398
总结
静电势作为连接电子结构理论与生物分子功能、材料界面行为的核心物理量,其计算方法已从量子力学的精确求解发展为涵盖多尺度建模的完整体系,当前研究正通过理论创新与技术升级不断拓展其应用边界。
从方法演化来看,静电势计算已形成多层次解决方案:量子化学框架下的DFT与PAW方法提供原子级精度,适用于小分子、固体表面等体系的电子结构与静电势关联分析。
连续介质模型通过介电常数与离子强度的引入,高效处理生物大分子在溶液中的静电势分布;多极子展开与可转移原子模型则以精度换效率,满足高通量筛选的需求。这种多层次方法体系确保了静电势计算能够适应从原子到宏观的全尺度研究。
当前研究趋势聚焦于三个方向:其一,机器学习加速高精度计算,图神经网络通过数据驱动的方式实现分子结构到静电势场的快速映射,将计算效率提升1-2个数量级,同时保持与DFT相当的精度,为药物设计、材料筛选等领域的大规模应用提供可能。
其二,非局部介电理论的普适化,通过引入空间色散效应修正传统PB方程在分子界面处的偏差,更准确描述氢键网络、溶剂化效应对静电势的调制,目前该理论已在蛋白–配体结合、膜蛋白功能等研究中展现出优势,未来需进一步拓展至复杂多相体系。
其三,静电动态耦合机制的解析,结合GPU并行化技术与分子动力学模拟,实时追踪蛋白质构象变化、材料相变过程中静电势的动态演化,例如在酶催化反应中,静电势的瞬时变化可揭示活性位点的质子转移路径,为催化机制解析提供新视角。
掌握静电势计算的核心模型与前沿工具,对于多个领域的理论研究具有不可替代的价值:在纳米材料设计中,可通过调控表面静电势优化其与生物分子的相互作用;在酶催化机制解析中,静电势的分布可揭示活性口袋对底物的选择性识别与活化机制;在药物开发中,静电势匹配度是评估药物分子与靶蛋白结合能的关键指标,直接影响药物的efficacy与安全性。
随着理论方法的持续完善与计算能力的提升,静电势计算将在连接微观电子结构与宏观功能性能的研究中发挥更核心的作用,推动从理性设计到实际应用的高效转化。