

说明:固态电池核心材料包括固态电解质(氧化物/硫化物/聚合物)、高镍三元/富锂锰基正极、硅基/锂金属负极。其中,硫化物电解质离子电导率达10⁻³~10⁻² S/cm,硅负极理论容量4200 mAh/g,锂金属负极容量3860 mAh/g。
DFT计算通过量化材料稳定性(如分解能)、离子输运机制(迁移能垒0.2 eV)及界面相容性(吸附能-1.8 eV),指导界面改性(如LiF@Li-Ag复合层),降低界面阻抗至1 Ω・cm²,推动固态电池性能突破。
固态电池的核心材料体系
固态电池的核心组件包括固态电解质、正极材料、负极材料,其材料选择直接影响电池的能量密度、安全性和循环寿命。以下是主流材料体系:
固态电解质
固态电池的核心材料体系由固态电解质、正极及负极构成,其性能取决于材料本征特性与界面协同作用。
固态电解质作为创新核心,氧化物体系(如Li₇La₃Zr₂O₁₂, LLZO)凭借Zr/La/Ti元素构建的三维锂通道实现高离子电导率(10⁻⁴~10⁻³ S/cm)及宽电化学窗口(>5 V),但机械刚性需界面优化;
硫化物电解质(如Li₁₀GeP₂S₁₂, LGPS)以S-Ge-P框架形成超离子导电路径,电导率达10⁻³~10⁻² S/cm(接近液态电解液),却易与水反应生成H₂S;聚合物体系(如PEO-LiTFSI)通过醚氧基与Li⁺配位传输离子,柔性优异但室温电导率仅10⁻⁶~10⁻⁵ S/cm,需纳米填料改性提升。
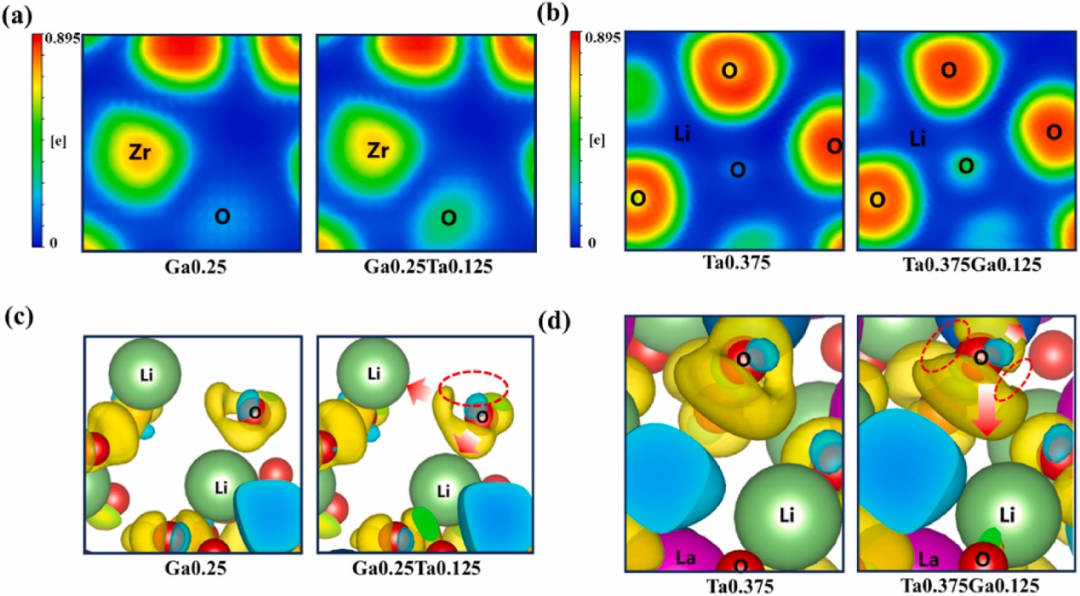

DOI:10.1016/j.jpowsour.2025.236287
正极材料
在固态电池正极材料体系中,高镍三元材料与富锂锰基材料展现出差异化的性能特征与发展潜力。
高镍三元材料(如 NMC/NCA 体系的 LiNi₀.₈Co₀.₁Mn₀.₁O₂)通过镍(Ni)、钴(Co)、锰(Mn)的原子级协同配比,在保持结构稳定性的同时显著提升能量密度 —— 其理论能量密度可达 250 Wh/kg 以上,为电动汽车长续航需求提供了关键材料支撑。
其中,高镍含量(Ni≥80%)是提升容量的核心驱动力,但也对材料循环过程中的晶格稳定性提出更高挑战。
富锂锰基材料(如 Li₁.₂Mn₀.₅Ni₀.₁Co₀.₂O₂)则凭借高锰(Mn)占比(≥50%)展现出显著的成本优势,锰元素的低成本与资源丰富性使其成为降低电池材料成本的重要方向,但其在充放电过程中存在电压衰减较快的固有缺陷,根源在于锰元素的价态波动引发的结构相变与过渡金属溶解。
当前,两类材料分别沿着“高能量密度 – 结构稳定性优化” 与 “低成本 – 电压衰减抑制” 的技术路径推进,前者聚焦表面包覆与元素掺杂以抑制镍离子混排,后者致力于通过氧晶格活化与缺陷工程调控锰的化学环境,共同推动固态电池正极材料向高性能、低能耗方向迭代升级。
DOI:10.1016/j.nanoen.2024.110276
负极材料
在固态电池负极材料领域,硅基负极与锂金属负极以超高理论容量成为研究焦点,却也面临亟待突破的技术瓶颈。
硅基负极(如 Si/C 复合材料)凭借 4200 mAh/g 的理论容量(约为传统石墨负极的 10 倍),成为提升电池能量密度的理想选择,但其在锂离子脱嵌过程中高达 300% 的体积膨胀会导致活性物质粉化与电极结构坍塌,制约循环寿命。
当前研究聚焦于纳米结构设计(如核壳结构、多孔硅骨架)与复合界面优化,通过碳基质的柔性缓冲作用缓解体积应力。
锂金属负极则以 3860 mAh/g 的理论容量和最低的氧化还原电位(-3.04 V vs 标准氢电极)被视为 “终极负极”,但其在充放电过程中易形成枝晶状锂沉积,刺穿电解质引发短路风险。
研究重点集中于界面工程(如人工固态电解质界面膜构建)与动力学调控(如优化锂离子通量分布),通过原子级模拟解析枝晶生长动力学,开发抑制枝晶的电解质组分与电极修饰策略。
两类材料分别代表“高容量 – 结构稳定性” 与 “高理论性能 – 安全性” 的技术路径,其突破依赖于材料化学、界面物理与计算模拟的交叉创新,有望为固态电池带来颠覆性能量密度提升与安全性革新。
DOI:10.1016/j.chphi.2023.100310
固态电池材料研究的核心
在固态电池材料研究中,密度泛函理论(DFT)通过原子尺度模拟材料电子结构与能量变化,成为精准指导材料设计的核心工具。
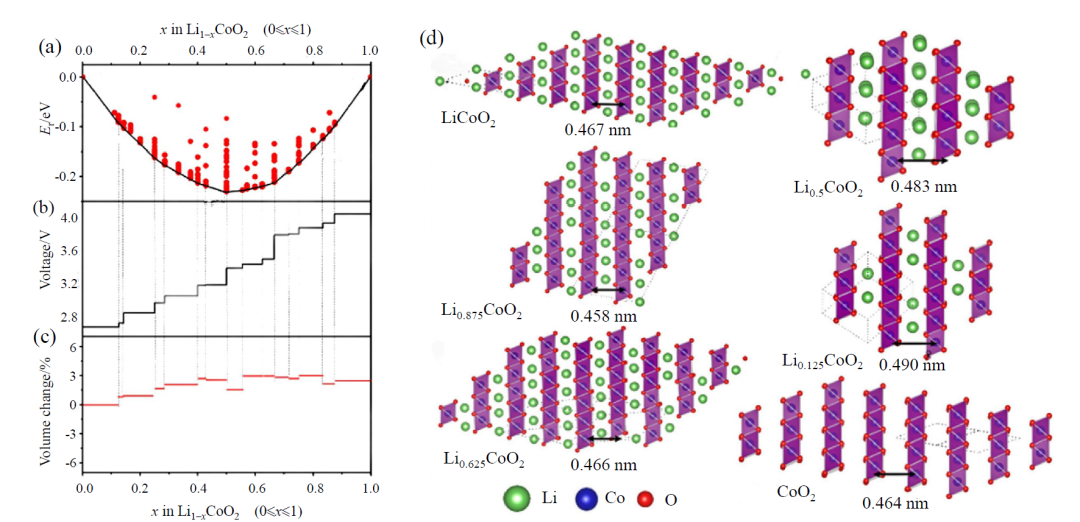
在材料稳定性预测方面,DFT 通过计算形成能与分解能量化材料热力学稳定性,例如对比层状与尖晶石相 LiCoO₂的分解能值,可提前预判充放电过程中的结构演变趋势,为抑制相变引发的容量衰减提供理论依据。
在离子输运机制解析中,DFT 通过模拟锂离子扩散路径与激活能(Ea)揭示传导动力学规律,如硫化物 Li₁₀GeP₂S₁₂中 Li⁺扩散激活能仅 0.2 eV,直接印证其高离子电导率的微观起源,为电解质离子通道优化提供 “原子级路线图”。
界面相容性优化则依赖于 DFT 对电极 / 电解质界面结合能与电子态密度(DOS)的计算,例如 LiCoO₂(003) 晶面与 LLZO (100) 晶面因弱结合能()导致界面电阻升高,此类计算可精准定位界面失效机制,指导界面修饰层的原子级设计。
电压平台预测方面,DFT 通过两相平衡体系的吉布斯自由能差(ΔG)推导理论电压,如 LiFePO₄/FePO₄体系计算值(3.4 V vs Li/Li⁺)与实验误差 ,为正极材料电压特性的精准调控建立可靠模型。
DFT 凭借对材料多维度性能的原子级解析能力,正推动固态电池研究从 “试错型实验” 迈向 “计算驱动型创新”,其与高通量筛选、机器学习的深度融合,将加速高稳定电解质、低阻抗界面等关键材料的开发进程,为固态电池的产业化突破奠定坚实的理论与技术基础。
DOI:10.12034/j.issn.1009-606X.223113
经典案例:硫化物电解质界面改性
在硫化物固态电池界面改性研究中,2023 年发表的《In-situ construction of Li-Ag/LiF composite layer…》一文通过 DFT(密度泛函理论)计算与实验验证的深度耦合,为解决锂金属与硫化物电解质的界面副反应提供了典型解决方案。
该研究聚焦于硫化物 Li₆PS₅Cl 与锂金属界面存在的枝晶生长与化学腐蚀问题,利用 DFT 从原子尺度解析界面改性机制:首先,通过计算 Li 原子在 Ag (111) 表面的吸附能(-1.8 eV),证实 Ag 单质对锂具有强亲和性,可作为锂沉积的高效成核位点,引导锂离子均匀沉积而非形成尖锐枝晶;
其次,计算 LiF 与 Li₆PS₅Cl 的界面形成能(-0.3 eV/Ų),发现 LiF 层与硫化物电解质间存在强相互作用,能够通过物理隔离效应阻断锂金属与硫化物的直接接触,抑制界面处的还原腐蚀反应。
基于上述 DFT 计算结果,研究团队筛选出 AgF 作为前驱体,通过原位反应在锂金属表面构建 Li-Ag 合金与 LiF 复合修饰层(LAF),该复合层兼具 “锂沉积引导” 与 “化学屏障” 双重功能:Li-Ag 合金通过低吸附能特性促进锂的均匀成核,LiF 层则凭借高界面形成能稳定锚定在电解质表面,形成机械强度高、电子绝缘性好的界面层。
实验数据表明,经 LAF 修饰的界面阻抗从传统体系的 > 100 Ω・cm² 骤降至 1 Ω・cm²,界面离子传导效率提升两个数量级以上,同时有效抑制了循环过程中锂枝晶的穿透行为。
该研究展现了 DFT 在界面工程中的核心价值 —— 通过量化吸附能、界面能等关键参数,实现从“改性思路提出” 到 “材料组分筛选” 的全流程理论驱动,为硫化物电解质与金属锂负极的界面兼容问题提供了可复制的解决方案,其方法论可推广至氧化物、卤化物等其他固态电池界面体系的设计,加速高安全性固态电池的产业化进程。
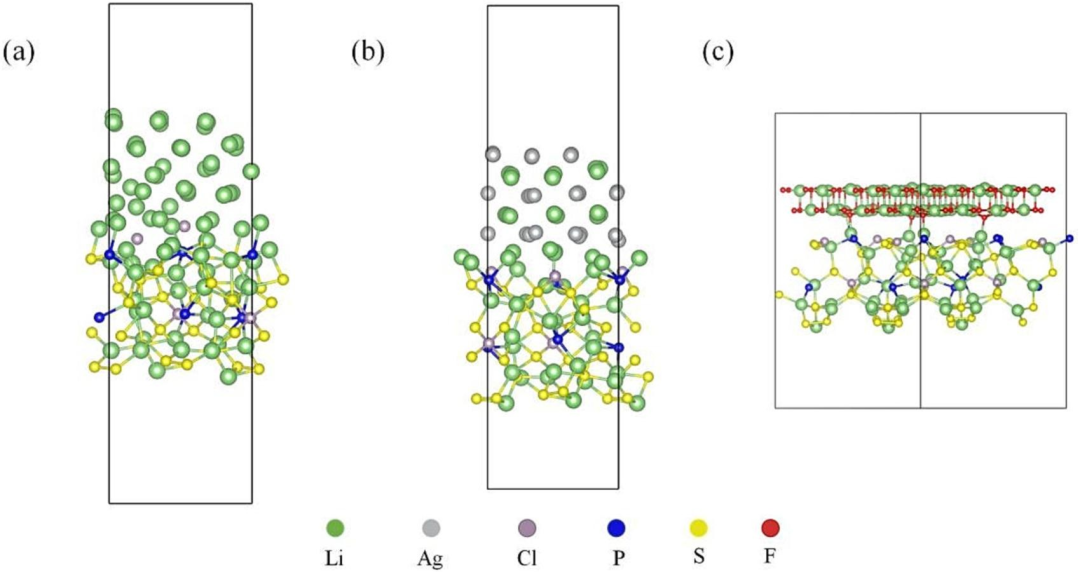
DOI:10.1016/j.cej.2023.147179
总结
密度泛函理论(DFT)作为固态电池材料研发的原子尺度解析工具,通过高通量筛选结合材料基因组数据库(如Materials Project)加速新型电解质发现(如硫银锗矿型Li₆PA₅X),机器学习则通过神经网络预测离子电导率等参数(如迁移能垒误差),大幅降低计算成本;
分子动力学(MD)与DFT联用实现动态界面模拟,揭示循环中枝晶生长应力(>1 GPa)与副反应路径(如Li₆PS₅Cl分解能垒0.8 eV),指导缓冲层设计(如LiF@Li-Zn复合层)。
DFT精准预测离子输运机制(如Li₃YCl₆低温活化能0.2 eV)、界面稳定性(界面能0.8 J/m²)及电压平台(误差),推动硫化物全固态电池临界电流密度提升至1.6 mA/cm²、循环寿命突破3000小时。
随着算法优化与算力升级,DFT将持续赋能固态电池从材料设计到产业落地的全链条创新。