

层状双氢氧化物(LDH)由二维层板与可调控层间阴离子构成,通过金属组合(如NiFe-LDH)及缺陷工程优化催化活性;羟基氧化物(如α-FeOOH)因晶型差异(层状/隧道结构)展现独特物化性质。
层状双氢氧化物(LDH)和羟基氧化物是两类在催化、能源存储和环境治理中广泛应用的功能材料。以下将从结构特征、分类体系及DFT理论计算应用三个维度展开深度解析。
LDH的结构与分类
基本结构特征
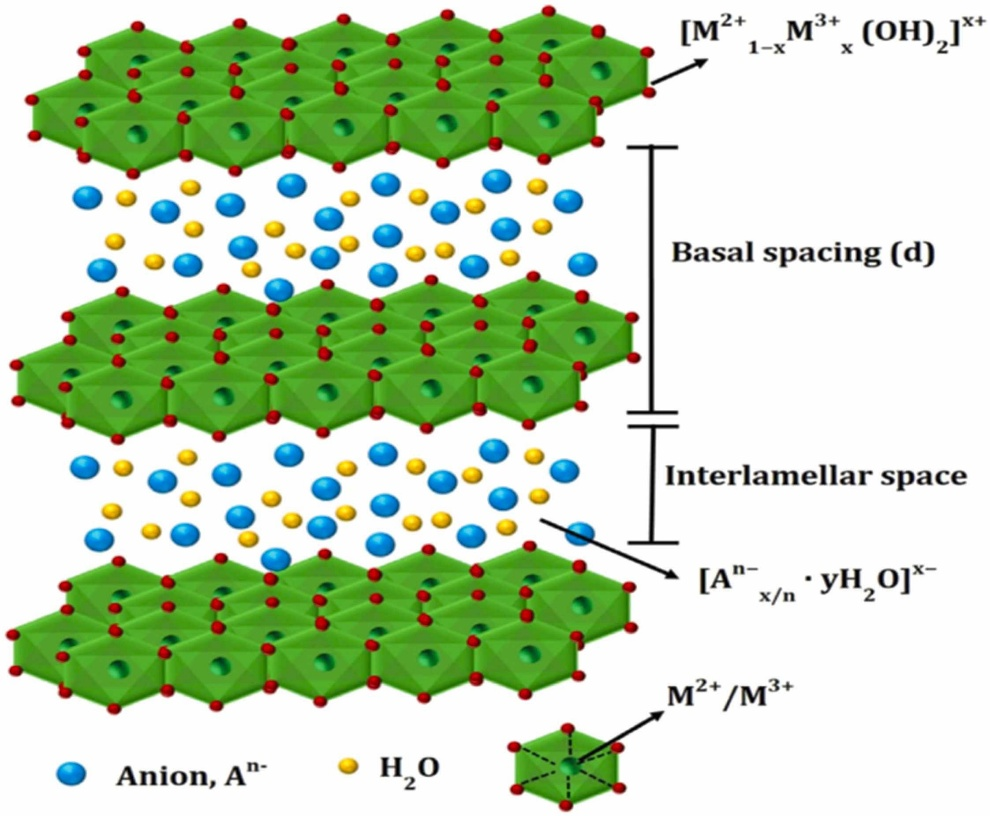
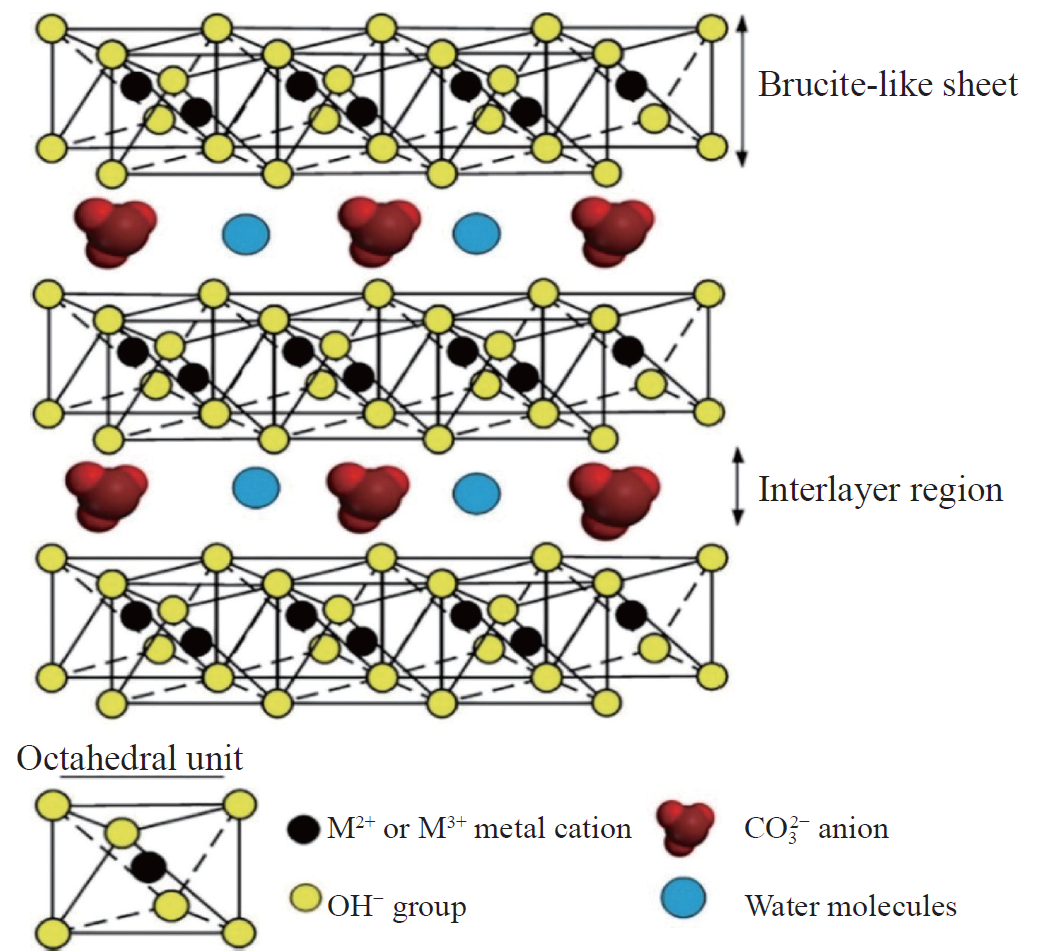
层状双金属氢氧化物(LDH)的化学通式为[M²⁺₁₋ₓM³⁺ₓ(OH)₂]^x+·[Aⁿ⁻]ₓ/ₙ·mH₂O,其结构由二维层板与层间域协同构成。
层板主体为二价(如Mg²⁺、Ni²⁺)与三价金属离子(如Al³⁺、Fe³⁺)通过羟基八面体共边连接形成的正电荷骨架,其中三价离子占比(x值)调控层板电荷密度(通常0.2≤x≤0.33);层间域通过静电作用容纳可交换的阴离子(如CO₃²⁻、NO₃⁻、Cl⁻)及水分子,形成动态平衡的夹层结构。
层板的正电荷(x+)由层间阴离子(Aⁿ⁻)补偿,其种类与排列方式(如单层或插层)直接影响LDH的热稳定性、离子交换能力及功能化潜力。
例如,CO₃²⁻因强静电作用难以被置换,而NO₃⁻可通过简单离子交换引入功能性阴离子(如MoO₄²⁻或有机磺酸根)。
LDH的分类基于金属离子组合(如MgAl-LDH、NiFe-LDH)与层间客体(如无机、有机或杂化阴离子),其结构可调性与高比表面积(>100 m²/g)使其在催化、吸附、药物载体及能源存储领域展现广泛应用前景。
 DOI:10.1016/j.apcatb.2023.123489
DOI:10.1016/j.apcatb.2023.123489
分类体系
层状双金属氢氧化物(LDH)的分类体系基于金属组成与结构特性,涵盖二元、三元、剥离型及复合结构等类型。
二元LDH(如MgAl-LDH、NiFe-LDH)由单一二价与三价金属构成层板,密度泛函理论(DFT)计算聚焦于层板电荷分布(如Al³⁺占比调控层板极性)与催化活性位点(如Fe³⁺的d轨道占据态);
三元LDH(如CuNiFe-LDH)通过引入第三金属(Cu²⁺)触发协同效应,DFT揭示其电子结构重构(如Cu的d带中心偏移)与多金属位点间的电荷转移机制。
剥离型纳米片(如CoAl-LDH单层)因高比表面积(>200 m²/g)与暴露活性位点,需通过DFT模拟表面吸附能(如CO₂吸附能-0.8 eV)及反应路径(如OER的O→OOH能垒);
三维复合结构(如LDH/碳纳米管)结合碳基体的导电性与LDH的离子交换能力,DFT分析界面电荷转移(Δρ=0.05 e/ų)及态密度耦合(如C的p轨道与LDH的d轨道杂化),指导高性能催化与储能材料设计。
这种分类体系与计算策略的结合,为LDH的功能化改性及跨领域应用提供了结构–性能关联的理论框架。
DOI:10.1016/j.jhazmat.2021.127612
DFT计算的典型应用
密度泛函理论(DFT)计算在层状双金属氢氧化物(LDH)的性能优化与机理研究中发挥关键作用。
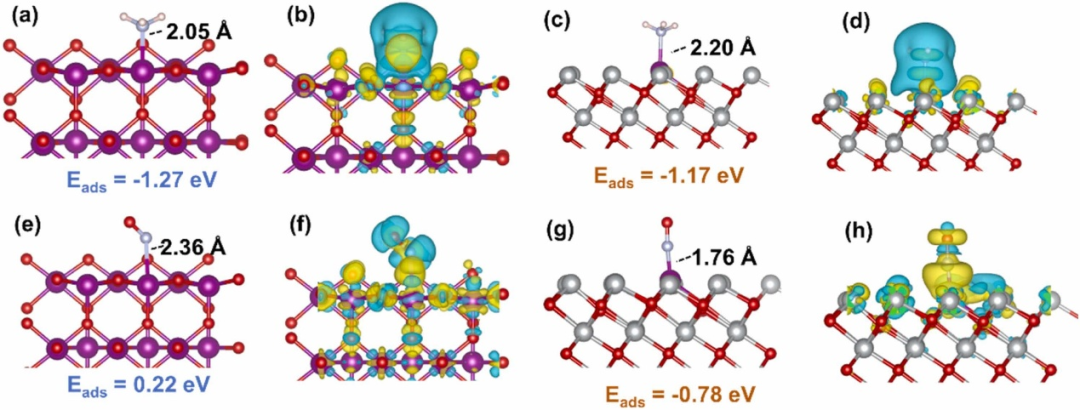
通过电子结构分析可解析层板金属的d带中心位置(如Ni²⁺的d带中心位于-2.1 eV),揭示其调控氧还原反应(OER)活性的内在关联——d带中心上移增强中间体吸附,优化*OOH形成能垒(0.45 eV);
层间阴离子效应研究中,DFT模拟NO₃⁻与CO₃²⁻的交换过程(吸附能从-1.5 eV调整至-0.8 eV),预测层间距膨胀(从0.76 nm增至0.89 nm)对传质效率与活性位点可及性的提升作用;
缺陷工程则通过空位形成能计算(如S掺杂空位形成能0.7 eV)筛选低能垒修饰策略,指导阳离子缺陷(如Fe空位)或阴离子掺杂(S替代O)设计,增强催化活性(如析氢反应过电位降低至0.12 V)。
这类计算从电子结构、层间化学到缺陷热力学多维度解析LDH的结构–性能关系,为高活性催化剂的设计与合成提供了原子级理论框架。
DOI:10.1016/S1872-2067(24)60215-9
羟基氧化物的结构与分类
基本定义
羟基氧化物(如FeOOH、GaOOH)是氢氧化物部分脱水的中间相,兼具氧化物与氢氧化物的结构特性,其基本结构由金属阳离子(M³⁺/M⁴⁺)与羟基(OH⁻)及氧离子(O²⁻)通过共边或共角八面体配位形成层状或链状骨架。
例如,α-FeOOH(针铁矿)为层状结构,羟基占据八面体顶点,而β-FeOOH(四方纤铁矿)呈隧道状排列,羟基位于棱边位置,结构差异导致其物化性质显著不同(如α相比表面积>150 m²/g,β相离子交换容量更高)。
根据羟基配位模式与晶体对称性,羟基氧化物主要分为α型(羟基顶点配位,单斜晶系)、β型(羟基棱边配位,四方晶系)及γ型(无序羟基分布,立方晶系),其中α型因稳定的层状结构在催化与储能领域应用广泛。
作为析氧反应(OER)催化剂,α-FeOOH通过Fe³⁺的d轨道与羟基的p轨道杂化优化中间体吸附能,而GaOOH则利用其宽禁带与表面羟基活性位点实现有机污染物的高效光催化降解。
这类材料的结构可调性与表面活性位点多样性,使其在能源转换与环境修复领域展现出重要潜力。
DOI:10.1016/j.mtchem.2021.100747
分类与晶型
铁基羟基氧化物的同质异构体(α、β、γ、δ型)因晶体结构与配位环境差异展现出独特的物化性质与应用潜力。
α-FeOOH(正交晶系,空间群Pbnm)的密度泛函理论(DFT)计算聚焦于氧空位形成能(如1.2 eV)及吸附位点竞争机制(如OH与OOH在Fe³⁺位点的吸附能差0.3 eV),解析其作为高效析氧反应(OER)催化剂的活性起源;
β-FeOOH(四方晶系,I4/mmm)通过Cl⁻掺杂调控电子结构,DFT模拟显示Cl⁻替代羟基可降低导带底(从-0.8 eV升至-0.5 eV),增强电导率并优化锂离子扩散路径(能垒0.4 eV→0.2 eV);
γ-FeOOH(三方晶系,R3m)的光生载流子迁移路径模拟揭示其沿[001]方向的快速输运特性(迁移率>200 cm²/(V·s)),支撑其在光催化降解有机污染物中的应用;
δ-FeOOH(六方密堆积,P63/mmc)的层间氢键网络通过DFT计算氢键能(~0.5 eV/键)与振动模式分析,阐明其对结构稳定性与离子嵌入/脱出可逆性的关键作用。
多晶型间的性能对比与DFT指导的改性策略(如缺陷工程、掺杂调控),为铁基材料在能源存储、催化及环境修复领域的精准设计提供了结构–功能关联的理论框架。
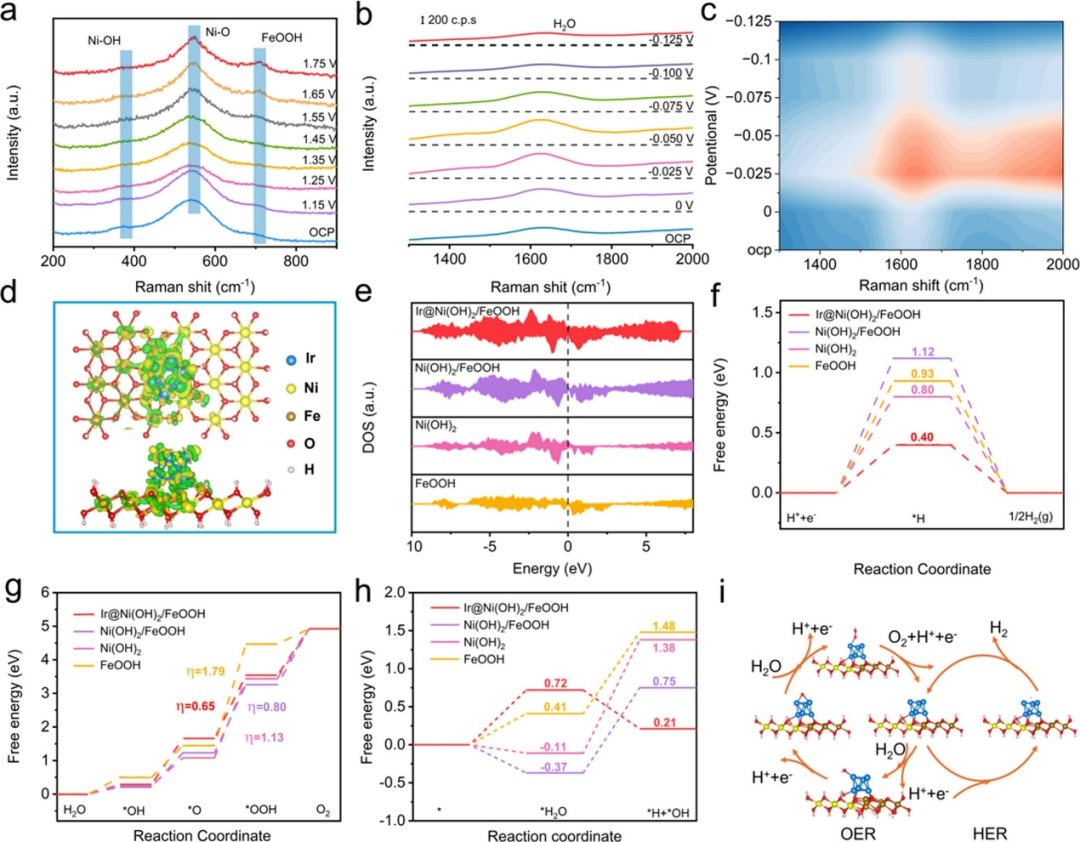
DOI:10.1016/j.apsusc.2025.163249
DFT计算的典型应用
在羟基氧化物材料的研究中,DFT 计算如同精准的 “原子级性能计算器”,为揭示材料本征特性提供多维度理论洞察。
在氧空位调控领域,通过计算不同晶型中氧空位形成能(如α-FeOOH 的 2.5 eV),研究者得以像 “缺陷工程师” 般精准设计空位分布,通过降低形成能优化表面吸附位点,为提升材料对反应中间体的捕获能力提供理论蓝图;
在电荷转移机制研究中,DFT 如同 “电子行为追踪仪”,通过模拟 Fe³⁺与 O²⁻间的电荷重新分布,清晰解析光催化过程中载流子分离的效率瓶颈 —— 电荷分布的不均匀性如何影响电子 – 空穴对的复合速率,进而为设计高活性光催化体系指明方向;
而在表面重构分析中,DFT 通过计算羟基化表面的吉布斯自由能,如同绘制反应活性的 “能量等高线图”,直观预测水氧化反应的活性趋势:低自由能路径对应的表面构型往往成为催化反应的 “高速通道”。
这一系列应用表明,DFT 不仅是连接宏观性能与微观机制的 “桥梁”,更通过定量计算将羟基氧化物材料的设计从 “试错模式” 带入 “精准调控时代”,为催化、能源转换等领域的突破奠定坚实的理论基石。
DOI:10.1016/j.apcatb.2025.125393
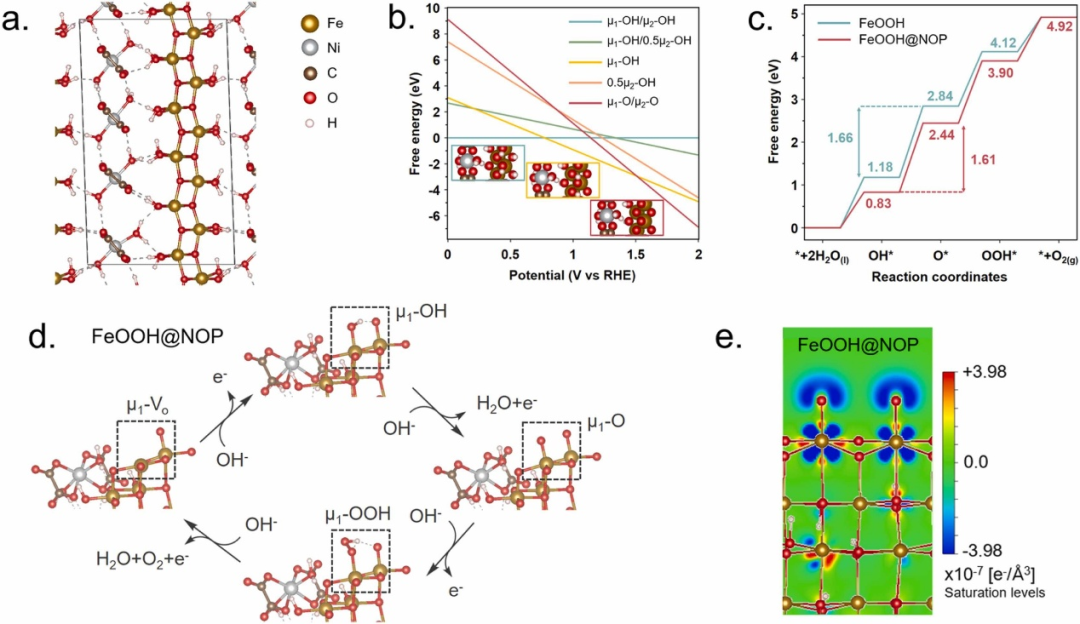
经典案例:NiFe-LDH
在《Promoting surface reconstruction of NiFe layered double hydroxide for enhanced oxygen evolution》这一研究中,DFT 计算如同精密的 “原子级显微镜”,为解析 S 掺杂 NiFe-LDH 的催化增强机制提供了多维度的理论支撑。
研究首先通过电子局域函数(ELF)分析,对 S 掺杂前后的材料电子结构进行 “精细画像”:对比图 1c-d 的 ELF 图谱可见,S 原子的引入打破了原有电子分布的局域状态,使电子离域程度显著提升,这一微观变化直接反映在脱氢能垒的优化上 —— 从原始结构的 4.05 eV 降至 3.74 eV,为后续催化反应的高效进行铺设了 “低能耗轨道”。
态密度(DOS)计算则像一把 “电子能量天平”,精准称量出掺杂对材料电子活性的调控效应。
图 1e-f 的 DOS 图谱显示,掺杂后费米能级附近的 d 电子态密度明显增加,这意味着材料表面活性位点对反应中间体的吸附 – 脱附能力得到优化,如同在催化反应的 “能量十字路口” 拓宽了快速通行的车道,为氧气析出反应(OER)的动力学加速提供了直接证据。
在反应路径模拟中,研究团队构建了 OER 过程的吉布斯自由能图,如同绘制了一幅催化反应的 “能量地形图”,清晰揭示了 S 掺杂带来的关键变革:未掺杂时,OOH 中间体的形成是整个反应的速率决定步骤(RDS),如同需要翻越陡峭的 “能量高山”;
而掺杂后,反应的速率控制步骤转变为 O 原子的脱附,对应的能量势垒显著降低,反应路径从 “攀登山峰” 变为 “跨越缓坡”。这种反应机制的转变,本质上是 S 掺杂诱导的电子结构调制与活性位点重构共同作用的结果,为实验中观察到的催化性能提升提供了原子尺度的合理解释。
这一系列 DFT 计算亮点不仅展现了理论工具在解析复杂催化体系中的强大威力,更通过定量分析与可视化表征,将“表面重构促进催化活性” 的宏观现象拆解为电子离域、态密度优化、反应路径调控等具体的微观机制,为设计高效 OER 催化剂提供了可复制的理论范式。
当理论计算与实验表征形成共振,NiFe-LDH 材料的催化潜力被进一步释放,这种 “计算驱动实验” 的研究模式,正成为推动能源催化领域突破的重要引擎。
DOI:10.12198/j.issn.1673-159X.5499
总结
在材料研究领域,研究者通过多尺度模拟并结合机器学习,高效加速高通量筛选金属组合与阴离子配比,为材料设计奠定基础;
借助分子动力学(MD)模拟,实时追踪层间水分子迁移对催化活性的动态影响,如同在原子级场景中观察反应的“实时直播”;还开发新泛函,精准量化 LDH / 氧化物异质结界面的电荷再分布情况,揭开界面效应的微观奥秘。
通过 DFT 理论计算,研究者如同拥有原子尺度的 “透视镜”,能深入解析材料的本征性质,为设计高性能 LDH 和羟基氧化物材料筑牢理论根基。
这一系列研究进展,正持续推动催化、能源存储等技术领域朝着更高效、更精准的方向革新,为相关领域的发展注入强劲动力。