

MXene材料通过表面工程(如缺陷调控、单原子掺杂)优化过硫酸盐(PMS)分解路径:单电子转移路径生成自由基(SO₄•⁻、•OH),非自由基路径主导单线态氧(¹O₂),多电子转移路径直接氧化污染物。
DFT计算揭示吸附能、能垒差异及电荷转移机制,验证Cu-SA/MXene的高选择性(99.7% ¹O₂)。结合机器学习预测界面特性,推动水处理催化材料的高效精准设计。



MXene材料的关键特性与活性位点


MXene材料(如Ti₃C₂Tₓ)作为二维过渡金属碳/氮化物,凭借其表面官能团(-O、-F、-OH)的可控修饰与独特电子结构,在催化与储能领域展现出显著优势。
其高导电性(~10⁵ S/m)源于金属性d轨道与碳/氮骨架的强杂化作用,为电催化反应(如析氢或氧还原)提供快速电子传输通道;
表面化学可调性通过缺陷工程(如氧空位形成能0.8 eV)或单原子掺杂(如Co/Cu锚定于Ti位点)优化活性位点的吸附构型,DFT计算表明Co@Ti₃C₂O₂可将过硫酸盐(PMS)的吸附能从-0.5 eV强化至-1.2 eV,显著提升自由基生成效率。
层间电荷分布通过插层(如Li⁺、NH₄⁺)或功能化(如氨基修饰)调控层间距(从0.98 nm扩展至1.25 nm),结合差分电荷密度分析揭示插层离子与MXene层板的电荷转移(Δρ=0.03 e/ų),增强离子扩散系数(如Na⁺扩散能垒降低至0.15 eV)与分子传质效率。
这类特性与活性位点设计的协同作用,使MXene在高级氧化、电催化及离子电池等领域成为高性能材料设计的理想平台。
DOI:10.1016/j.cocom.2025.e01051
PMS分解的三大路径与DFT台阶图机理
单电子转移路径
MXene材料(如Cu-SA/MXene)通过单原子过渡金属位点(如Cu)催化过硫酸盐(PMS)分解的单电子转移路径,其核心机制为PMS分子末端O原子吸附于活性位点,引发O-O键断裂并生成硫酸根自由基(SO₄•⁻)和羟基自由基(•OH)。
密度泛函理论(DFT)计算显示,Cu单原子锚定显著增强PMS的吸附能(-2.04 eV),远高于未修饰MXene(-0.8 eV),且差分电荷密度分析证实电子从Cu的d轨道向PMS的O原子转移,导致O-O键长从1.45 Å拉伸至1.62 Å,削弱键合强度。
自由能台阶图进一步揭示反应路径的动力学特征:PMS吸附步骤(ΔG = -1.8 eV)为决速步,而后续O-O键断裂的能垒仅0.3 eV,表明该路径具备高效的反应驱动力。
通过结合吸附构型优化、电子转移分析及能量演化模拟,DFT从原子尺度阐明了Cu-SA/MXene的催化活性起源,为精准设计高活性过硫酸盐活化材料提供了理论范式,推动其在有机污染物降解等环境修复技术中的实际应用。
DOI:10.1021/acscatal.3c03303
非自由基路径(单线态氧¹O₂为主)
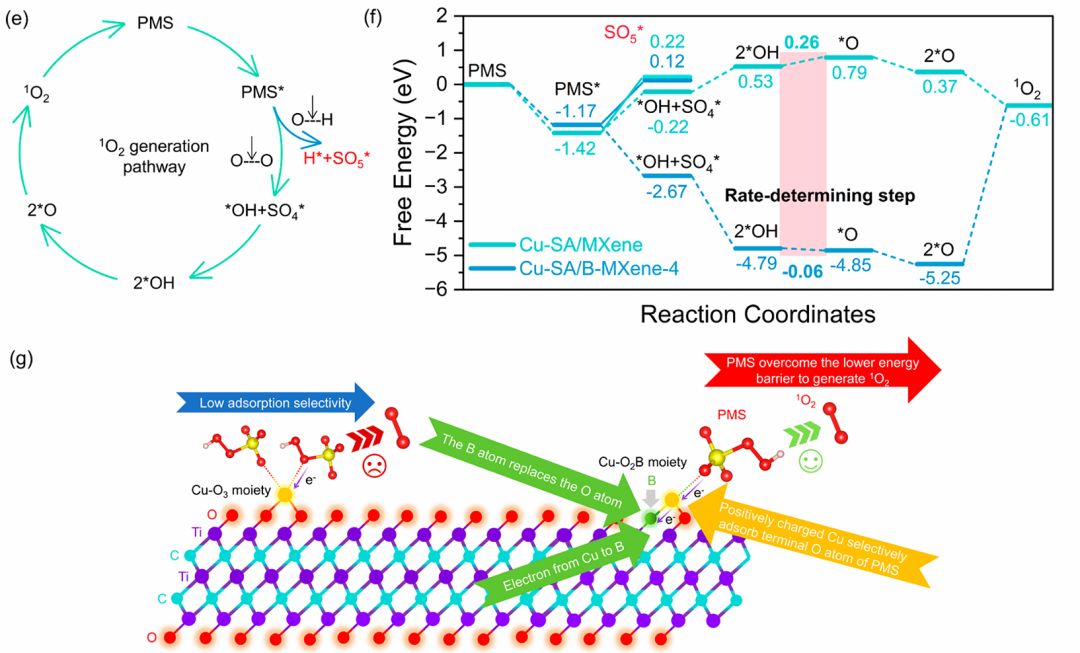
MXene材料(如Cu修饰的Ti₃C₂Tₓ)在催化过硫酸盐(PMS)分解的非自由基路径中,通过弱电子转移机制主导单线态氧(¹O₂)的生成。
该路径的核心步骤为PMS在MXene表面发生自分解,形成中间体SO₅•⁻,随后通过歧化反应转化为¹O₂。
密度泛函理论(DFT)计算表明,MXene表面Cu单原子的3d轨道与PMS的O 2p轨道发生杂化(图5c),使PMS的最低未占轨道(LUMO)能级降低1.2 eV,增强电子接受能力,促进弱电子转移(ΔQ=0.3 e⁻)。
同时,SO₅•⁻中间体在MXene表面的形成能比液相中低0.7 eV,表明材料表面显著稳定中间体并促进其积累。自由能台阶图显示,SO₅•⁻→¹O₂的转化能垒仅为0.5 eV,远低于自由基路径的1.2 eV(图5f),证实非自由基路径在动力学上的显著优势。
这种电子结构调控与中间体稳定化的协同作用,使MXene基催化剂在选择性生成¹O₂的同时抑制副反应,为高级氧化工艺中难降解有机污染物的定向去除提供了高效催化体系的设计策略。
DOI:10.1021/acscatal.3c03303
多电子转移路径(直接氧化)
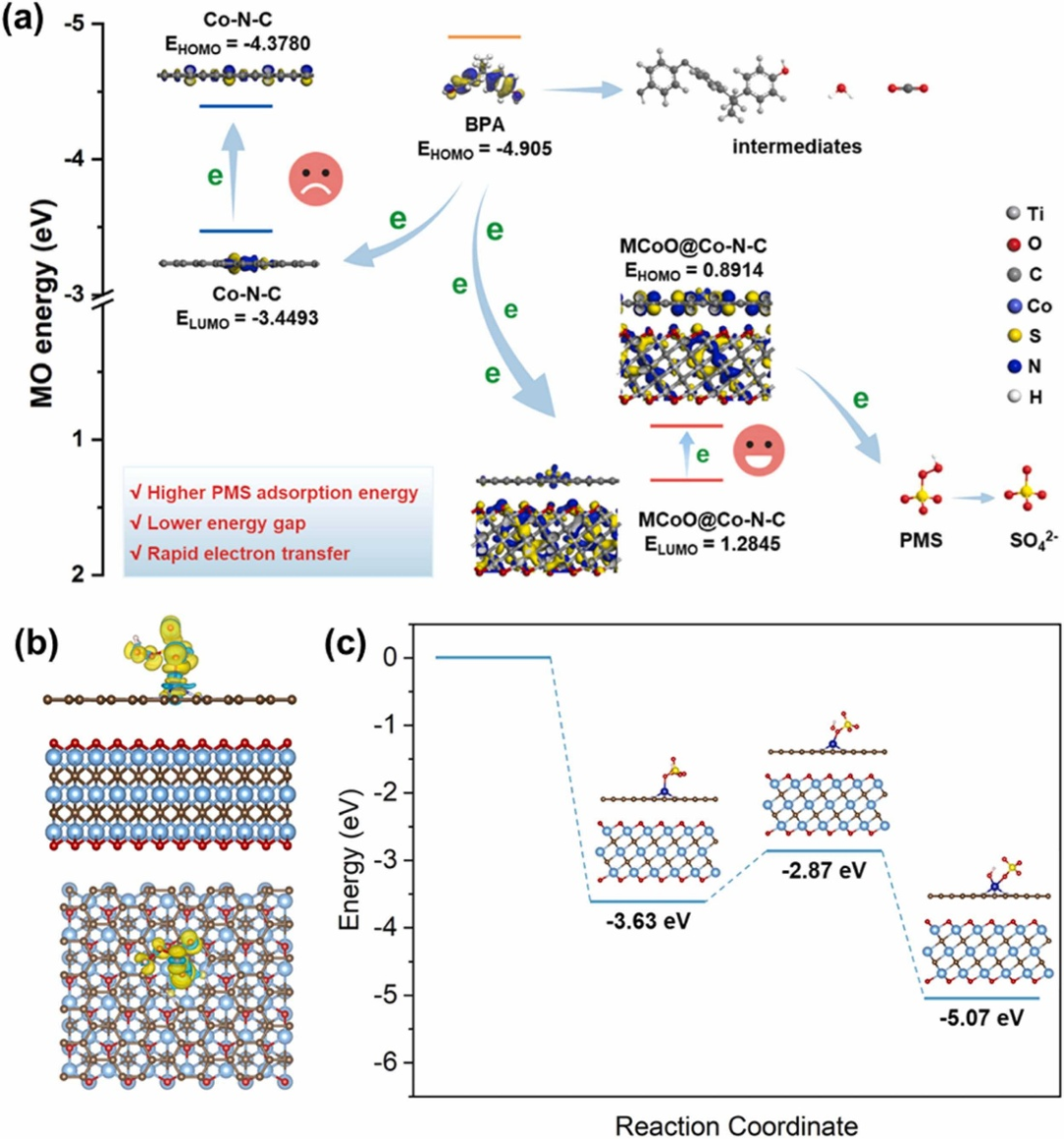
MXene材料(如Ti₃C₂Tₓ)在过硫酸盐(PMS)分解的多电子转移路径中,通过导电层介导富电子污染物(如双酚A,BPA)的直接电子转移,形成亚稳态复合物(MXene-PMS*),实现污染物的高效氧化。
密度泛函理论(DFT)计算表明,BPA的最高占据分子轨道(HOMO,-5.85 eV)与MXene的最低未占轨道(LUMO,-4.30 eV)的能级差较小(1.55 eV),显著促进电子隧穿效率;
界面电荷差分分析进一步揭示,MXene-PMS*复合物中电子在界面处局域化(Δρ=0.05 e/ų),形成“电子池”效应,增强PMS的氧化能力。该机制通过多电子协同转移直接活化PMS,绕过自由基生成的中间步骤,避免副反应对目标污染物的竞争消耗。
MXene的高导电性(~10⁵ S/m)与表面官能团(如-O、-OH)的调控作用,不仅优化电子传输路径,还通过静电作用稳定PMS吸附构型(吸附能-1.2 eV),为复杂体系中污染物的定向降解提供了高效、低能耗的催化策略。
这种直接氧化路径的揭示为设计非自由基主导的高级氧化工艺奠定了理论基础,推动MXene基材料在水处理与环境污染修复中的实际应用。
DOI:10.1016/j.apcatb.2022.122136
经典案例:PMS-MXene体系
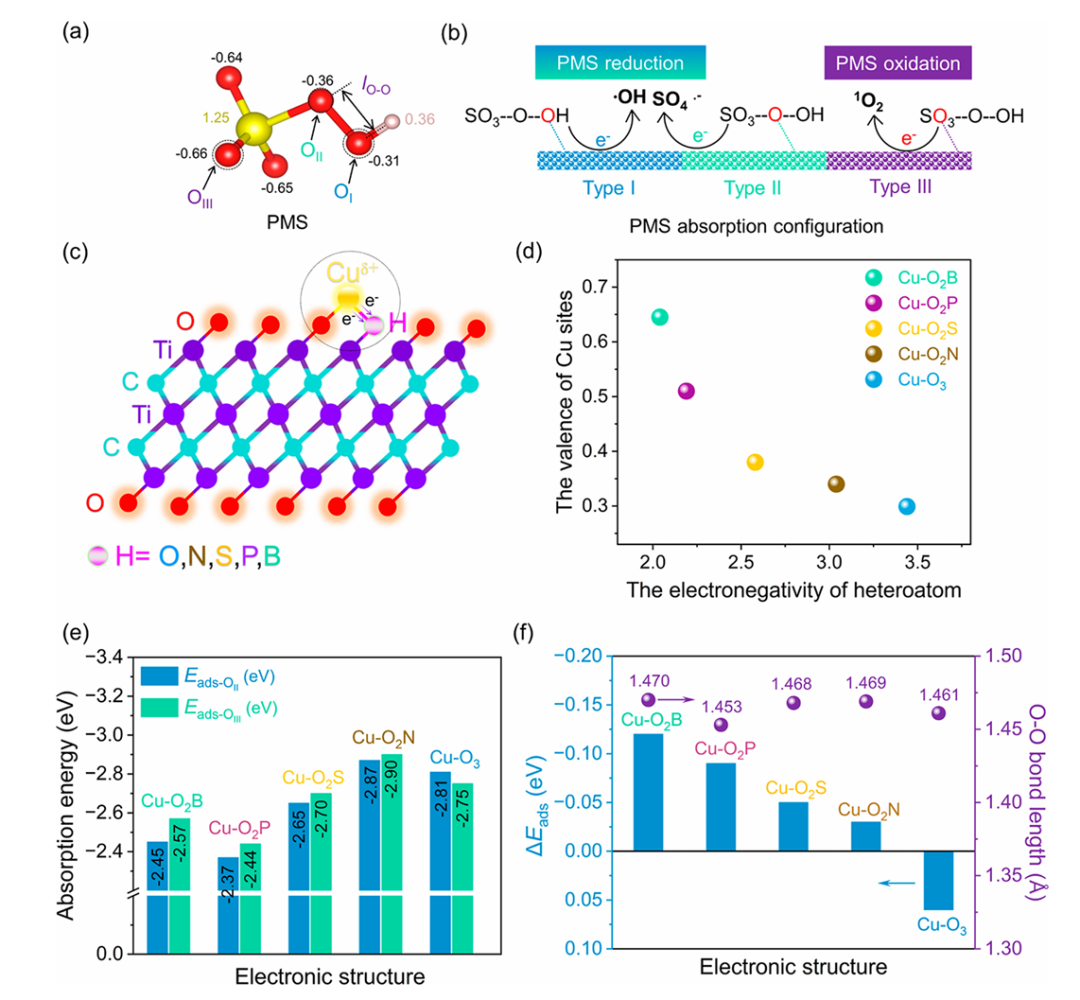
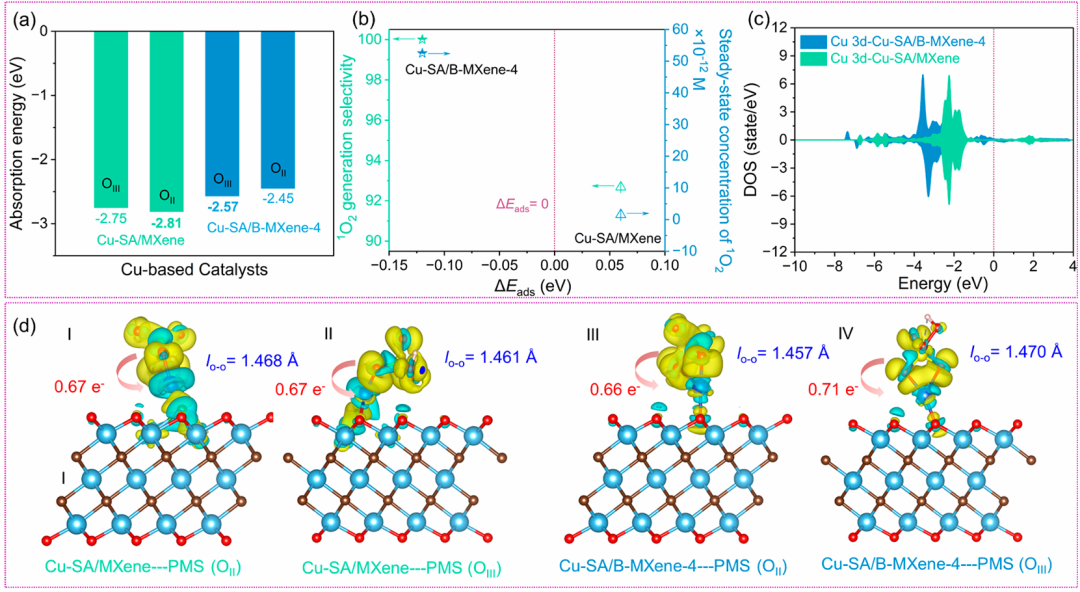
在 Liu 等人发表于《Applied Catalysis B: Environmental》2023 年的研究中,DFT 计算如同精密的 “原子级反应解码器”,为揭示 Cu 单原子 / MXene 体系活化过硫酸盐(PMS)的高选择性机制提供了多维度的理论洞察。
通过吸附构型优化,研究者发现 PMS 在 Cu-O₂B 位点的吸附能低至 – 2.42 eV,显著低于未掺杂位点的 – 1.70 eV,这一量化差异直观展现了 B 掺杂对 PMS 结合力的强化作用 —— 更低的吸附能如同为 PMS 分子设置了更稳定的 “锚定点”,使其更易被活化。
结合图 5d 的差分电荷分析,黄色电子积累区域集中于 PMS 的末端 O 原子,表明该位点成为优先活化的 “热点区域”,为后续反应路径的定向调控埋下伏笔。
反应路径模拟通过势能面揭示了催化选择性的核心奥秘:¹O₂生成路径的能垒比 SO₄・⁻路径低 0.7 eV,这一能量优势直接解释了实验中高达 99.7% 的 ¹O₂选择性。
两种路径的分岔源于活性位点的微结构差异 ——反应模型显示,Cu-O₃位点倾向于生成 SO₄・⁻,而 Cu-O₂B 位点则通过 SO₅・⁻中间体导向 ¹O₂生成,如同在反应的 “能量十字路口” 设置了明确的导向标,使反应沿着更低能耗的路径进行。
这种精准的路径控制,本质上与 B 掺杂引发的电荷重分布密切相关:Bader 电荷分析表明,B 掺杂使 Cu 位点电子密度降低(电荷态 + 0.15 e),形成更缺电子的活性中心,这一变化如同为 PMS 分子开启了 “氧化模式”,促进其作为氧化剂而非还原剂参与反应,从电子结构层面杜绝了副反应路径的竞争。
这些 DFT 计算亮点不仅从吸附能、能垒差异、电荷状态三个维度构建了完整的催化机制图谱,更通过与实验数据的高度契合(如选择性结果),验证了理论模型的可靠性。
当吸附构型的微观优化、反应路径的能量 Landscape、电荷分布的量子调控形成协同效应,Cu 单原子 / MXene 体系的高活性与高选择性不再是实验现象的孤立呈现,而是被拆解为可追溯、可量化的原子级作用机制。
这种 “计算驱动催化” 的研究范式,不仅为单原子催化剂的理性设计提供了可复制的方法论,更让环境催化领域的反应机理研究从“宏观推测” 进阶到 “原子级精准解析”,为高效降解污染物的催化材料开发开辟了新的理论赛道。
DOI:10.1021/acscatal.3c03303
总结
在环境催化领域,MXene 材料通过表面工程手段(缺陷调控、元素掺杂、单原子锚定)如同拥有 “分子级方向盘”,可精准调控过硫酸盐(PMS)的分解路径,实现对催化反应的定向控制。
其中,高导电性 MXene(如 Ti₃C₂O₂)凭借优异的电子传输能力,更易通过单电子转移路径激活 PMS,生成具有强氧化能力的自由基物种;
若在 MXene 表面构建弱电子受体位点(例如引入 B 掺杂的 Cu 单原子),则能导向非自由基路径,通过能量更优化的电子结构设计,促使 PMS 以 ¹O₂等非自由基形式参与反应,显著提升催化选择性;
而多电子转移路径的高效运行,依赖于污染物分子与 MXene 表面的能级精准匹配,如同为不同污染物 “量身定制” 反应通道,实现氧化降解的精准对接。
这种基于表面工程的路径调控策略,不仅揭示了 MXene-PMS 体系的催化本质,更开启了 “按需设计” 催化材料的新范式。
未来研究若能结合机器学习算法,对 MXene-PMS 界面的电子结构、吸附能等关键特性进行高通量预测,便可像 “催化材料裁缝” 般,针对特定污染物的降解需求,精准设计具有最优界面特性的 MXene 材料,推动水处理技术向高效、低碳、个性化的方向迈进。
这种理论与数据驱动的协同创新,正为环境催化领域铺设一条从 “经验试错” 到 “精准定制” 的升级之路,为解决复杂水质污染问题提供更可持续的解决方案。