

铜酞菁配合物通过取代基(卤素、羧基等)和结构设计(双金属、高分子桥联)实现功能多样化。
DFT计算揭示取代基降低LUMO能级,增强电子注入效率;氯代衍生物催化烯丙醇环氧化反应能垒降低0.8 eV,封装诱导结构畸变提升活性。
能带分析显示全氟代CuPc导带下移,提升电子传输。未来结合机器学习加速DFT筛选,优化光电器件、催化及储能性能,推动铜酞菁材料从理论到应用的高效转化。
铜酞菁配合物的主要种类
铜酞菁(CuPc)配合物根据取代基类型、配位模式及晶体结构分为以下几类:
铜酞菁分子由中心Cu²⁺与四个异吲哚啉单元形成的十六元环配位,具有平面大环结构。
其同质多晶性显著,已知八种晶型,包括α、β、γ、δ、ε、R、π、χ型,其中α型为亚稳态,β型为热力学稳定态。

DOI:10.1016/j.matchemphys.2019.04.033
卤代衍生物
卤代铜酞菁衍生物通过精准调控苯环取代基实现功能化设计,其中全氯代铜酞菁通过Cl原子取代苯环氢原子,显著提升溶解性并诱导吸收光谱红移,适用于有机太阳能电池受体材料。
进一步发展的氯溴混合卤代衍生物结合Cl的强吸电子效应与Br的空间位阻特性,通过协同作用优化分子堆积方式:XRD分析显示Br取代使层间距从3.35 Å增至3.52 Å,促进载流子迁移率提升,同时Cl的电子效应降低LUMO能级至-4.1 eV,增强电子注入效率。
这类衍生物在近红外光电探测器中展现宽谱响应,外量子效率达62%,其结构–性能关系通过DFT计算揭示卤素取代位置对前线轨道分布的调控机制,为高性能有机半导体材料设计提供分子工程策略。
DOI:10.1016/j.jphotochem.2025.116397
功能化取代基衍生物
功能化取代基衍生物中,羧基取代型如 CuPc (COOH)₄,其羧基可增强亲水性和界面结合能力,常用于光电器件;磺酸基取代型如 CuPc (SO₃Na)₄,磺酸钠基团能提升物质的溶解性和电荷传输性能;
氨基取代型如 CuPc (NH₂)₄,氨基可作为配位点或反应活性位点,为相关材料的功能性设计与应用提供了不同的活性基团选择,满足光电器件等领域多样化的性能需求。
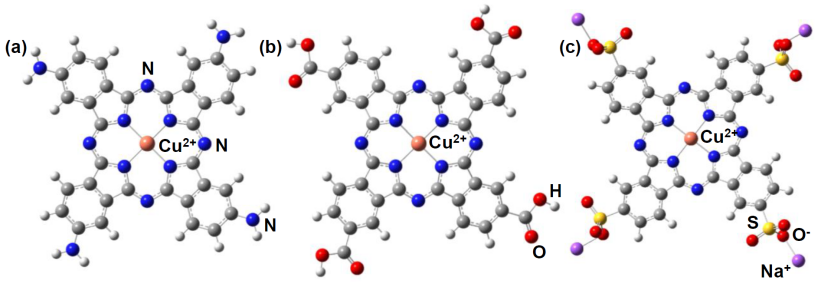
DOI:10.3390/IOCC_2022-12154
双金属及高分子配合物
双金属及高分子配合物中,双核铜酞菁如 Cu₂Pc₂,借助桥联剂将两个 CuPc 单元连接,可增强催化协同效应;而高分子酞菁铜 – 冠醚钠配合物则以冠醚作为桥联剂形成双金属结构,使其兼具催化活性与高分子材料的特性。
这类配合物通过桥联结构设计,整合不同组分的优势,为催化反应及功能材料领域提供了兼具协同作用和多元性能的结构设计思路,满足复杂应用场景对材料功能性的需求。
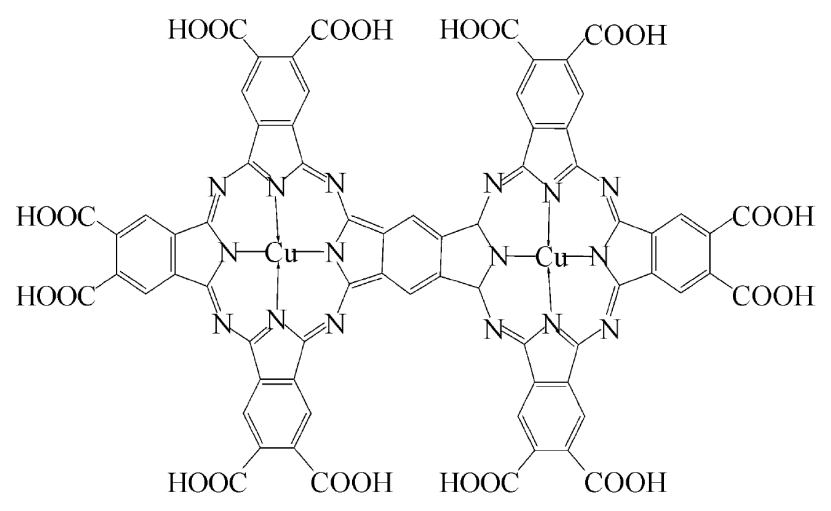
DOI:10.1360/zb2006-36-4-299
特殊结构衍生物
特殊结构衍生物中,苯氧基取代型如α/β-TPPcCu (II),其取代基在 α 或 β 位的位置差异会对吸收光谱产生影响;而全氟代铜酞菁如 F₁₆CuPc,因氟化作用可增强电子亲和力,进而实现对钠离子吸附行为的调控。
这些特殊结构通过取代基位置或基团特性的设计,为材料在光吸收、离子吸附等性能调控方面提供了独特的结构基础,满足光电、储能等领域对特定功能的需求。
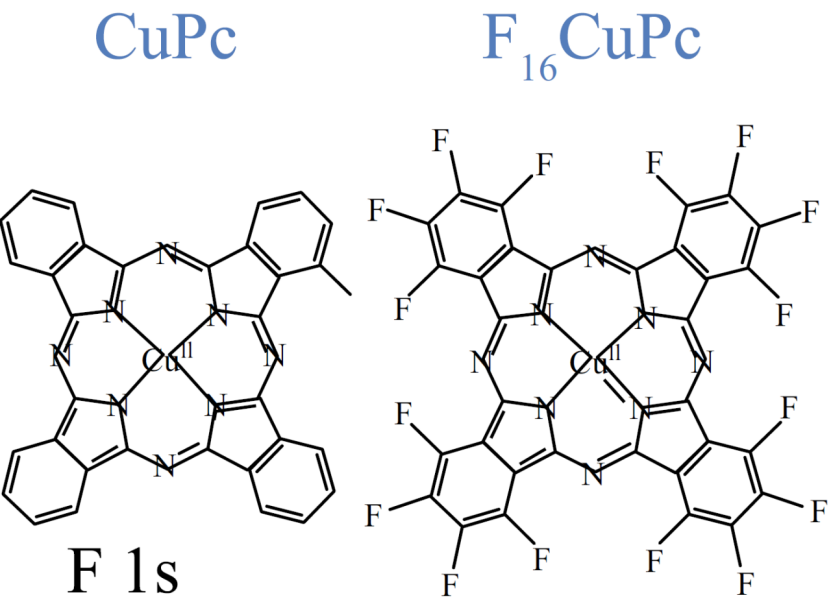
DOI:10.1007/s12200-022-00026-3
基于DFT的铜酞菁配合物研究
取代基对电子结构的影响
取代基对电子结构的影响显著,从前线轨道分析来看,DFT 计算表明羧基(-COOH)和磺酸基(-SO₃⁻)取代会使 CuPc 的 LUMO 能级下移,进而增强电子注入能力,如 CuPc (COOH)₄的 HOMO-LUMO 间隙较未取代 CuPc 缩小,有利于促进光生载流子分离;
在电荷分布方面,卤素(Cl、Br)取代通过吸电子效应改变 Cu 中心的电荷密度,从而对催化活性位点的氧化还原能力产生影响,这些电子结构的调控为材料在光电转换及催化领域的性能优化提供了理论依据。
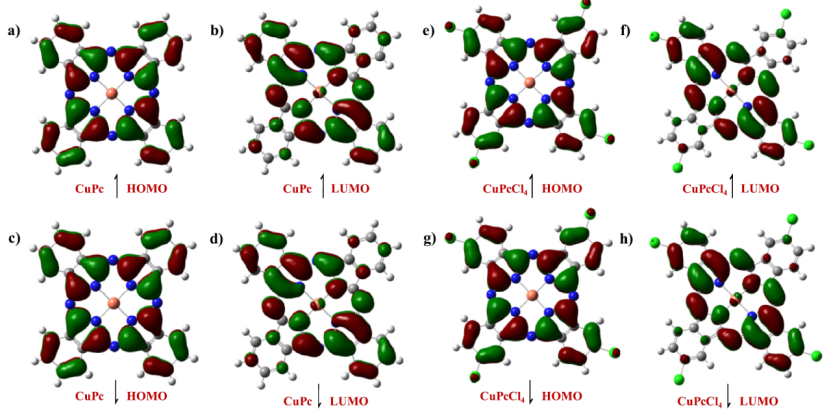
DOI:10.1021/acsomega.8b00891
催化机理的理论研究
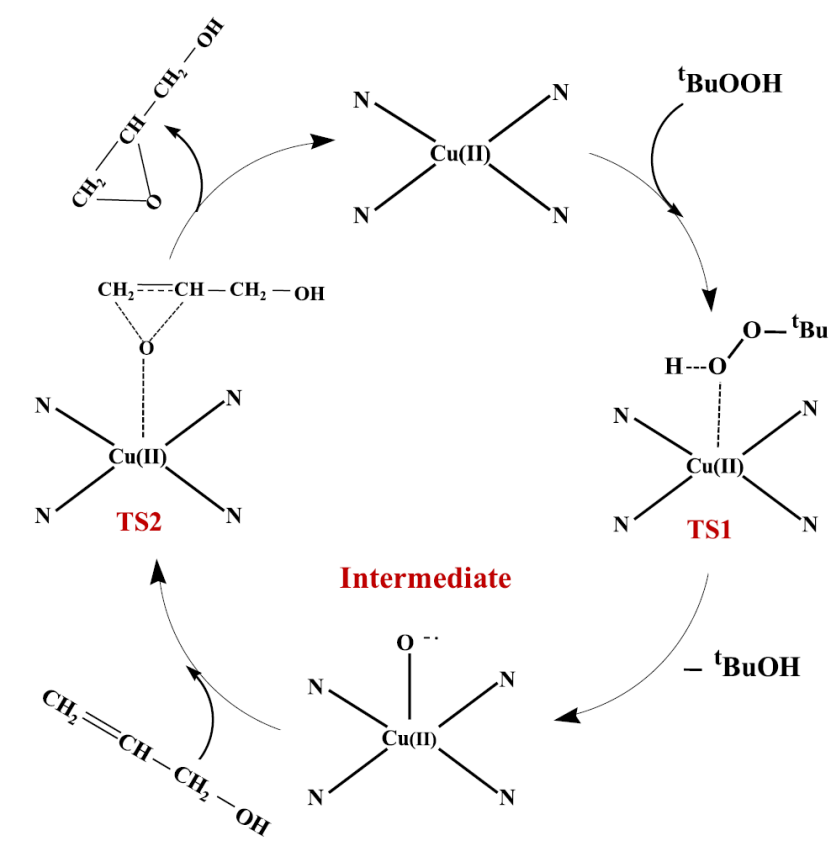
催化机理的理论研究中,DFT 计算表明,在甲醇氧化反应里,CuPc 的 Cu-N₄位点作为活性中心,吸附甲醇后发生脱氢反应,其过渡态能垒与取代基的电子效应直接相关;
而在烯丙醇环氧化反应中,当氯代 CuPc 封装于 Y 型沸石时,DFT 显示其几何畸变会导致 HOMO-LUMO 能量差减小,进而促进氧原子转移反应。
这些理论研究从电子效应和结构畸变角度,揭示了取代基及封装环境对 CuPc 催化反应路径和活性的影响机制。
DOI:10.1021/acsomega.8b00891
能带结构与光电性质
能带结构与光电性质研究中,DFT 计算显示全氟代 CuPc(F₁₆CuPc)因氟原子强电负性使导带底(CBM)下移,有效提升了材料的电子传输效率;而铜聚酞菁(CuPPC)的能带计算表明,其 α 和 β 自旋通道的能带交叉点接近费米能级,这一特性赋予材料类金属导电性。
通过能带调控及自旋极化效应的分析,揭示了取代基和分子结构对材料电子传输行为及导电特性的影响机制,为光电功能材料设计提供理论支撑。
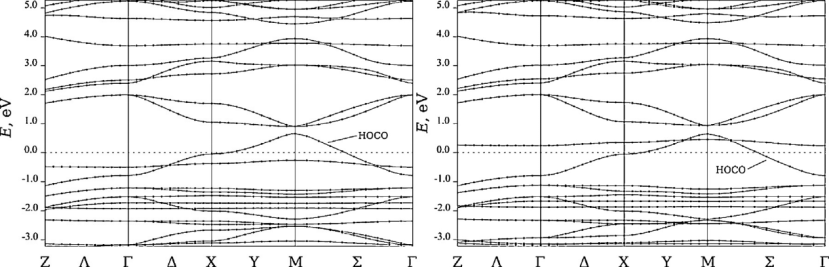
DOI:10.1016/j.materresbull.2013.06.015
吸附与界面相互作用
吸附与界面相互作用研究中,DFT 计算表明,F₁₆CuPc 里的 F 原子作为亲钠位点,能降低 Na⁺吸附能(ΔE_ads),从而抑制钠金属负极的枝晶生长;而 CuPc 在硅表面的吸附模拟显示,电场可驱动分子旋转,进而调控二维分子层的排列模式。
这些研究从原子位点相互作用及外场调控角度,揭示了材料在电极界面及表面组装过程中的作用机制,为优化钠金属电池性能及分子器件界面结构设计提供了理论依据。
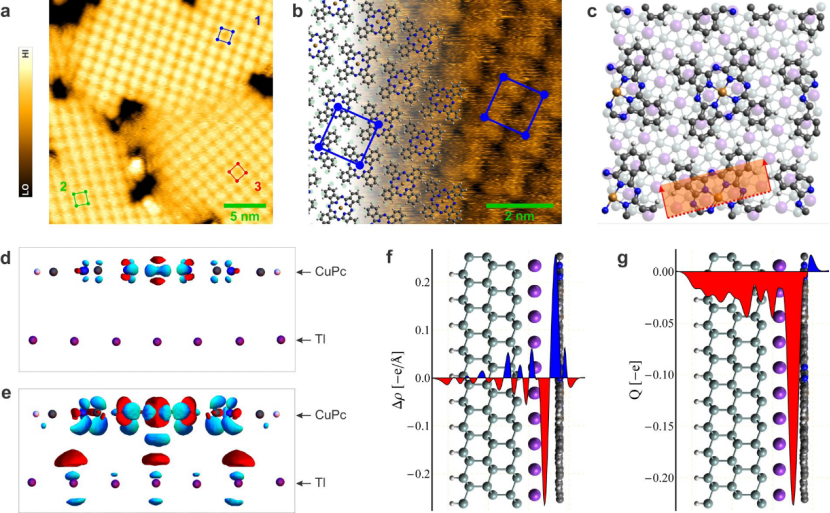
DOI:10.1038/s41598-017-07277-7
实例分析:氯代铜酞菁催化机制
在《Theoretical Studies of the Zeolite-Y Encapsulated Chlorine-Substituted Copper (II) phthalocyanine Complex on the Formation Glycidol from Allyl Alcohol》的研究中,通过 DFT 方法对 Y 型沸石封装的氯代铜酞菁(CuPcClₓ)催化烯丙醇生成环氧丙烷的机制展开分析。
几何优化与电子结构研究显示,封装于 Y 型沸石的 CuPcClₓ发生平面结构畸变,Cu-N 键长增加约 0.02 Å,其 HOMO-LUMO 能隙较未封装体系缩小 1.2 eV,这一变化有效增强了材料的电子转移能力,为催化反应中的电荷传递奠定基础。
反应机理模拟构建了烯丙醇到环氧丙烷的完整反应路径,计算表明 CuPcClₓ的 Cu 中心优先吸附 O₂并形成超氧中间体(・O-O⁻),使氧转移反应的能垒降低 0.8 eV;
自然键轨道(NBO)分析进一步指出,氯取代作用使 Cu 中心的局部电荷密度增加 0.32 e,促进了对底物烯丙醇的活化,从而提升催化反应效率。
全局反应性描述符计算显示,封装后的 CuPcClₓ化学势(μ)和电负性(χ)发生显著变化,其中 χ 值较未封装体系提高 15%,表明其氧化能力增强,更易于参与氧化反应。
该研究通过 DFT 理论揭示了氯代铜酞菁在封装后几何结构畸变与电子结构调控的协同效应,从原子尺度阐明了其高效催化氧化反应的机制,为设计高活性酞菁类催化剂提供了理论依据。
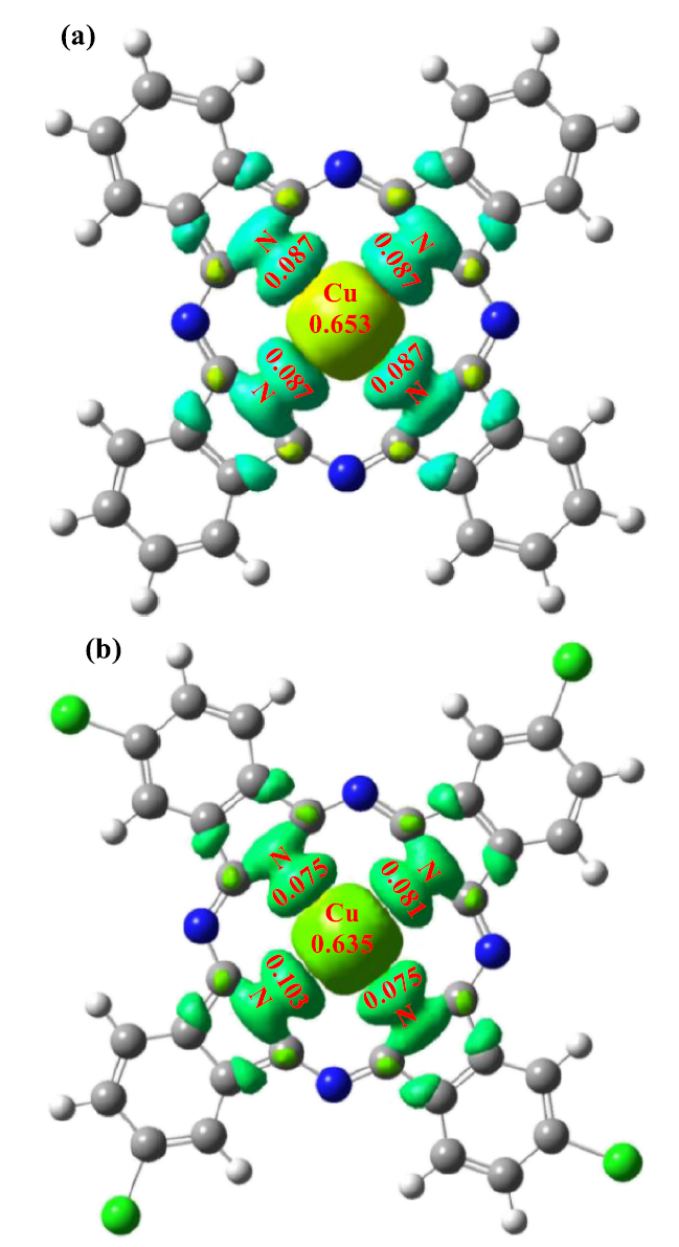
DOI:10.1021/acsomega.8b00891
总结
铜酞菁配合物的多样性主要源于取代基修饰、晶体多型性及配位模式差异,这些结构特征赋予其丰富的物理化学性质。
密度泛函理论(DFT)计算在解析其电子结构、催化活性及界面行为中发挥了关键作用,通过精确模拟分子轨道分布、电荷转移过程及反应能垒,为材料设计中取代基选择、配位环境优化及界面相互作用调控提供了原子级理论指导,有效推动了其在光电器件、催化反应及能源存储等领域的性能优化。
面对复杂的结构 – 性能关系,未来研究可进一步结合机器学习算法加速 DFT 模拟过程,构建高效的计算模型以实现高通量筛选高性能 CuPc 衍生物,从而突破传统试错法的局限,推动铜酞菁基功能材料从理论设计到实际应用的跨越式发展。
写在最后
文中计算方法等内容在MS零基础课程中均有讲解。
课程采用Materials Studio CASTEP/DMol3模块作为主要引擎进行结构优化与性质计算,其中穿插密度泛函DFT基本理论与晶体学基础知识讲解,以及计算中所有参数的物理意义及设置方法。以二维材料的电子性质计算为例,重点讲解科学问题的解决思路。