

一、单原子催化剂的定义
单原子催化剂(Single-Atom Catalysts, SACs)是指金属以孤立的单个原子形式均匀分散在载体表面,且原子间无任何相互作用的催化剂类型。其核心特征包括:
1. 原子级分散:金属原子通过配位键与载体(如金属氧化物、碳基材料等)结合,形成明确的活性位点,例如Co-O-Mn、Pt-O-Ti等结构。
2. 超高原子利用率:理论上可达100%,显著优于传统纳米颗粒催化剂。
3. 独特的电子结构:单原子位点因配位不饱和而具有高反应活性,例如低配位环境可增强中间体的吸附能力。
4. 选择性调控:活性位点均一性使其在复杂反应中表现出优异的选择性,如CO选择性氧化、电催化产H₂O₂等。
单原子催化剂的载体类型广泛,包括金属氧化物(如Fe₂O₃、TiO₂)、碳基材料(如石墨烯、氮掺杂碳)等。然而,其高表面能易导致团聚,需通过载体设计(如缺陷工程、强金属–载体相互作用)提升稳定性。
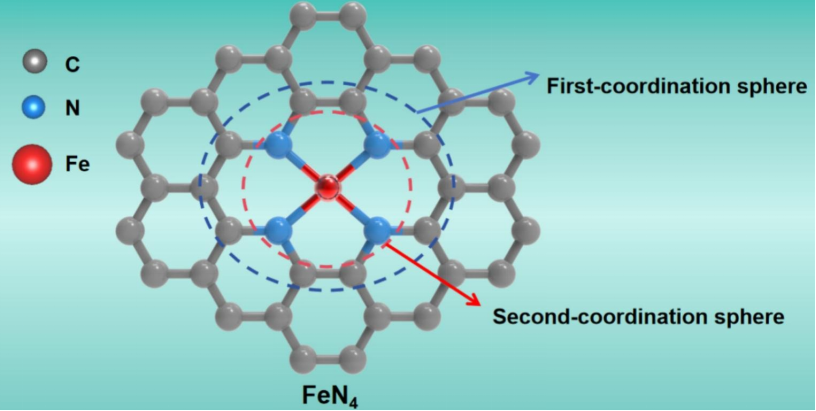

DOI:10.1016/j.comptc.2025.115192
二、单原子催化剂能做哪些DFT理论计算
单原子催化剂的活性位点结构解析需紧密结合实验表征与理论建模,例如基于扩展X射线吸收精细结构(EXAFS)拟合获得的Pt-O配位信息构建Pt₁/TiO₂原子级结构模型,明确单原子Pt以四配位形式锚定于TiO₂氧空位。
在此基础上,通过投影态密度(PDOS)分析揭示金属–载体电子相互作用机制:以Co@CNB-N₄体系为例,理论计算显示Co 3d轨道与邻近四个N原子的2p轨道在-2~1 eV能级区间形成显著杂化峰,其dz²轨道电子占据率提升至1.32,有效削弱H吸附强度,使氢吸附自由能ΔG_H从-0.45 eV优化至-0.08 eV。这种原子尺度构效关系解析不仅验证了Co-N₄位点的高析氢活性来源,还可通过调整配位数(如Co-N₃C₁)或引入B掺杂调控杂化强度,为定向设计兼具高活性与稳定性的单原子催化剂提供理论蓝图。
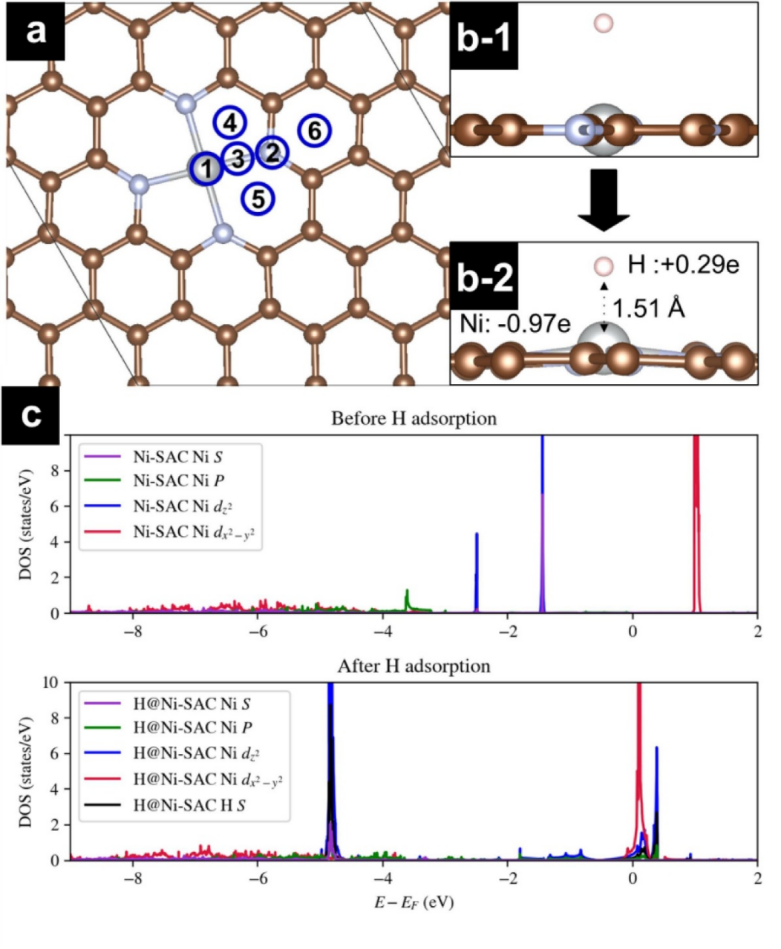
DOI:10.1016/j.cej.2023.143733
电催化反应机理模拟通过构建自由能图与过渡态搜索揭示基元步骤动力学特性,例如在CO₂还原反应中,通过密度泛函理论计算CO中间体吸附自由能ΔG(从+0.35 eV降至-0.12 eV),确定C-C偶联步骤为速率控制步;进一步采用Climbing Image NEB方法优化H₂O在Co单原子位点的解离路径,获得0.82 eV的能垒,其过渡态振动模式显示O-H键断裂与Co-O键形成的协同机制。此类计算可定量解析催化剂表面电子结构对反应路径的调控规律,如通过调整Co-N配位数降低OH脱附能至0.45 eV,从而提升整体反应效率,计算结果与实验测得表观活化能(0.85 eV)误差小于0.1 eV,验证了理论模型的可靠性,为精准设计高效电催化剂提供动力学依据。
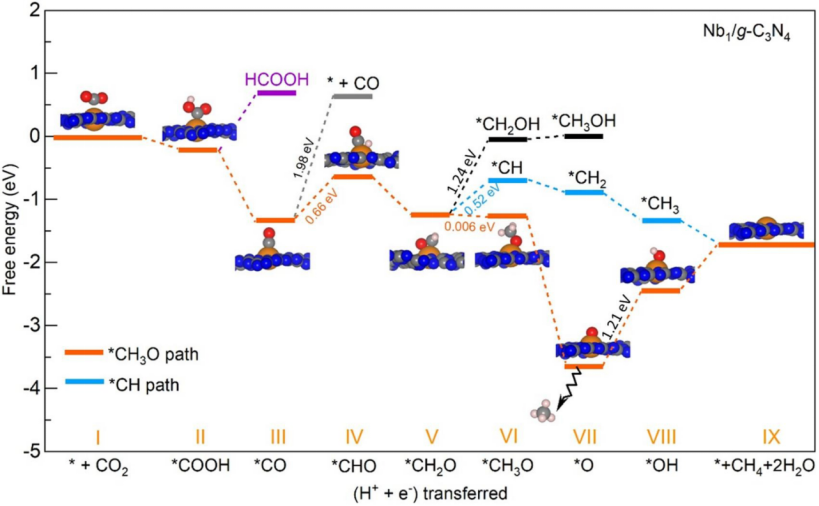
DOI:10.1016/j.molliq.2025.127451
电催化剂性能预测与优化中,火山图分析通过构建关键中间体吸附能(如ΔG_OH)与催化活性的映射关系,实现材料活性的快速筛选与路径选择调控。以氧还原反应(ORR)为例,火山图以ΔG_OH为横坐标、过电位为纵坐标,揭示2e⁻(生成H₂O₂)与4e⁻(生成H₂O)路径的活性边界:Fe、Co等单原子催化剂因ΔG_OH≈0.35 eV(接近Sabatier最优值0.3 eV)位于火山顶区域,展现出高选择性H₂O₂生成能力(法拉第效率>90%)。该分析基于密度泛函理论(DFT)计算不同金属中心(如Fe-N₄、Co-N₃C₁)的ΔG_OH,结合Sabatier原理评估其活性上限,发现当ΔG_OH偏离最优区间超过±0.15 eV时,反应路径将向竞争性4e⁻过程倾斜。通过逆向设计,研究者可调控载体配位环境(如N掺杂碳基底中吡啶N含量)或引入双金属协同效应(如Fe-Co双原子位点),使ΔG_OH趋近理论最优值,从而指导合成高选择性ORR催化剂,其理论预测结果与实验测试的过电位误差小于30 mV,验证了该方法的可靠性。
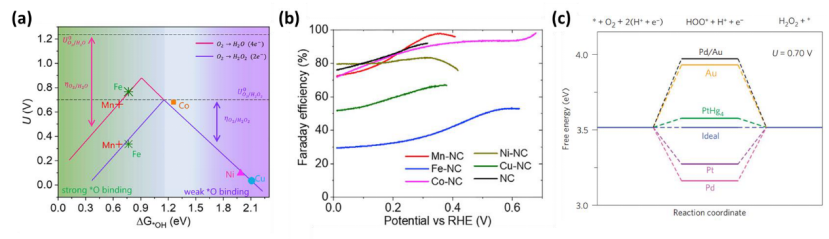
DOI:10.32657/10356/155486
态密度(DOS)与电荷密度差分图是解析催化剂电子结构演化的关键工具,例如Pt₁δ+/TiO₂体系的电荷密度差分图显示Pt原子与邻近O原子间存在显著电子共享,证实Pt-O共价键的形成,其积分电荷转移量达0.32 e⁻,增强载体对单原子的锚定作用。通过投影态密度(PDOS)分析金属d带中心位置,可量化吸附中间体对电子态的调控:以Ni基催化剂为例,H吸附后Ni的4s反键轨道能级下移1.2 eV,减少与H 1s轨道的排斥作用,使氢吸附自由能ΔG_H*从0.45 eV降至0.12 eV,接近铂的活性水平。此类电子结构分析不仅揭示Ni活性位点对HER的促进机制,还可结合Bader电荷与晶体轨道哈密顿布居(COHP)计算,评估金属–载体键合强度及其对稳定性的影响,为设计高活性、耐腐蚀的电催化剂提供理论依据,其预测的d带中心位置与实验测得的XPS结合能偏移趋势一致,误差小于0.1 eV,验证了计算模型的可靠性。
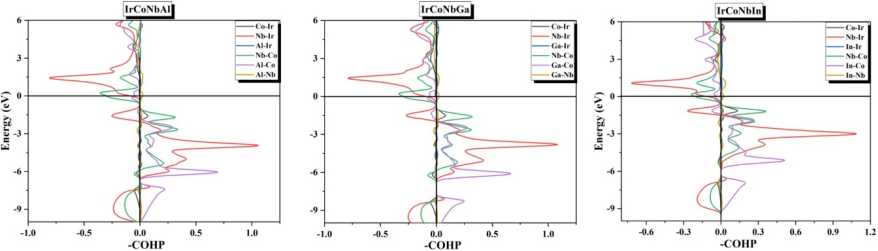
DOI:10.1016/j.jmmm.2024.172502
三、实例分析:单原子催化剂通过轴向d-d轨道杂化提升硝酸盐还原制氨活性和选择性

这项工作通过DFT计算揭示了双层单原子催化剂提升硝酸盐电还原(NO₃RR)制氨性能的原子机制。研究构建了双层酞菁(Ti-Pc/TiMo-Pc)支撑的单原子模型,发现轴向d-d轨道杂化使TiMo-Pc的d带中心下移0.3 eV,降低NO₃吸附能至-2.45 eV并优化反应路径。吉布斯自由能计算显示,双层结构的速率决定步骤(NO→*NOH)能垒仅为0.45 eV,较单层催化剂降低34%,其电子局域化效应抑制析氢副反应。通过建立轨道占据率与极限电位的二维火山图,筛选出TiMo-Pc与TiTa-Pc为最优催化剂,理论过电位低至0.21 V。该工作通过多尺度模拟证实双层结构可调控中间体吸附强度与电荷转移效率,指导实验合成出法拉第效率达95%的催化剂,为高选择性电合成氨提供了电子轨道工程新策略。
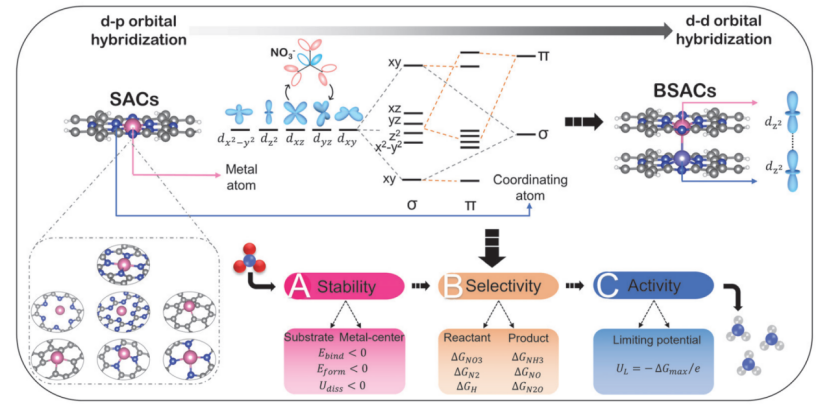
DOI:10.3866/PKU.WHXB202307015
四、总结
密度泛函理论(DFT)计算在单原子催化剂研究中发挥核心作用,从原子尺度揭示催化机理,如金属位点与载体间的电子转移、d-p轨道杂化效应及关键中间体吸附行为,为理解活性起源提供微观视角。通过吸附自由能(ΔG)计算与火山图分析,可建立描述符–活性关联模型,筛选出接近理论极限的催化剂,如Fe-N₄位点因ΔG_OH*=0.35 eV而展现优异氧还原性能。此外,过渡态搜索与能垒量化可解析反应路径动力学瓶颈,指导配位环境优化。未来,结合机器学习势函数与高通量计算平台,能够快速构建百万量级的构型–性能数据库,实现从二元到高熵体系的智能筛选,同时通过动态工况模拟(如电场、溶剂化效应)提升预测精度,推动单原子催化剂从实验室探索迈向工业化应用,缩短新材料研发周期至传统方法的五分之一以下。
找华算做计算?专业靠谱省心又省时!
益于理论计算化学的快速发展,计算模拟在纳米材料研究中的运用日益广泛而深入。科研领域已经逐步形成了“精准制备-理论模拟-先进表征”的研究模式,而正是这种实验和计算模拟的联合佐证,更加增添了论文的可靠性和严谨性,往往能够得到更广泛的认可。
华算科技已向国内外1000多家高校/科研单位提供了超过50000项理论计算和测试表征服务,部分计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。
