

TiO2光催化的定义和基本原理
TiO₂光催化是一种利用光能激发半导体材料(如TiO₂)产生电子-空穴对,从而引发氧化还原反应的技术。其基本原理是:当TiO₂吸收波长小于或等于其禁带宽度(通常为387.5nm)的光子时,价带中的电子被激发到导带,形成电子-空穴对。电子(e⁻)在导带中移动,而空穴(h⁺)在价带中留下。这些电子和空穴分别与吸附在TiO₂表面的物质(如水分子H₂O和氧气O₂)发生反应,生成具有强氧化能力的羟基自由基(·OH)和超氧阴离子自由基(·O₂⁻)。这些活性自由基能够降解有机污染物并将其矿化为无害物质,如二氧化碳和水。
TiO₂光催化具有高效性,其电子 – 空穴对分离效率高、寿命长,利于促进氧化还原反应;具有安全性,化学稳定性好、无毒无害且无二次污染;具有广谱性,对多种有机物和无机物污染物均有效,能利用紫外光甚至可见光反应。并且,TiO₂光催化技术广泛应用于环境净化(废水处理、空气净化)、能源转化(水分解制氢)以及抗菌灭菌等领域。

(https://doi.org/10.1002/wcms.1686)
2020年由 Zhuo Jiang等人在Nature中发表了一篇关于TiO2光催化的文章。他们在铬对苯二甲酸酯基 MOF(MIL-101)及其衍生物的不同孔隙内生长 TiO₂,制备了 TiO₂-in-MOF 复合材料,利用多种表征手段研究了 TiO₂在 MOF 中的位置、数量、分布情况以及复合材料的光催化性能。
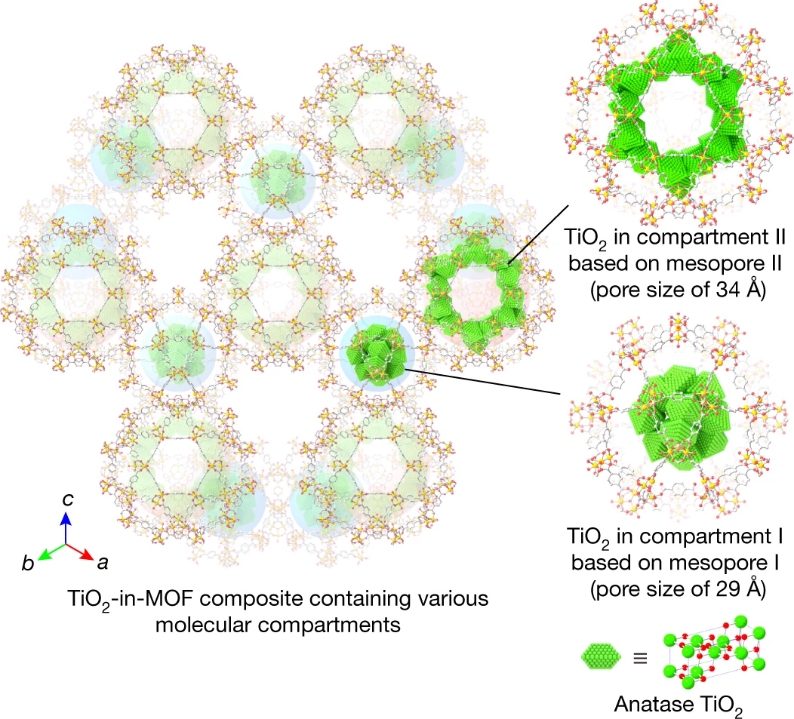
(Nature586,549–554 (2020). https://doi.org/10.1038/s41586-020-2738-2)
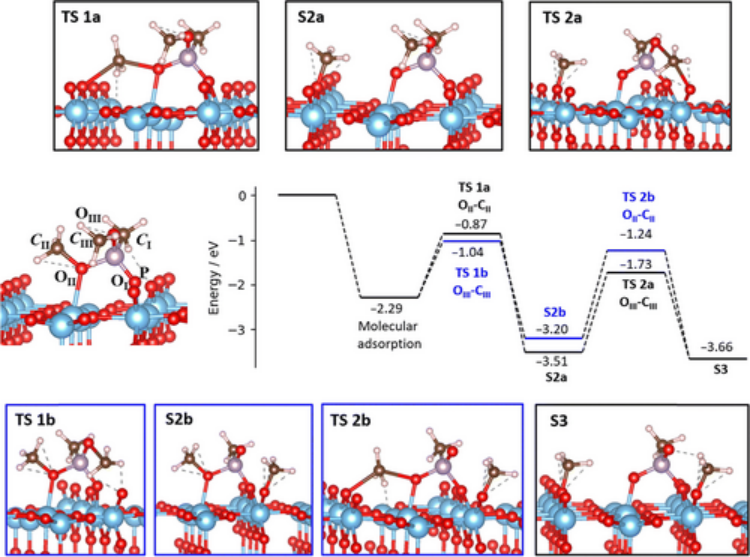
研究发现,TiO₂主要以锐钛矿形式存在于 MOF 的介孔中,且随着 TiO₂含量增加,其在不同介孔中的分布呈现特定规律。在 CO₂光还原测试中,42%-TiO₂-in-MIL-101-Cr-NO₂样品性能最佳。理论计算部分,运用密度泛函理论(DFT)计算来确定 MIL-101 系列中金属簇中间体状态的能级,从热力学角度深入了解反应过程。研究发现,在不同金属的 SBUs 中,只有 Cr³⁺/Cr²⁺和 Fe³⁺/Fe²⁺氧化还原对所在的 SBUs 为电子从 TiO₂转移到 MOFs 提供了合理途径,而 Al³⁺/Al 的氧化还原电位不合适,不利于电子转移,这与 TAS 测量中含 Al 的 TiO₂-in-MOF 样品中 TiO₂单元激发电子寿命较短的结果相符。
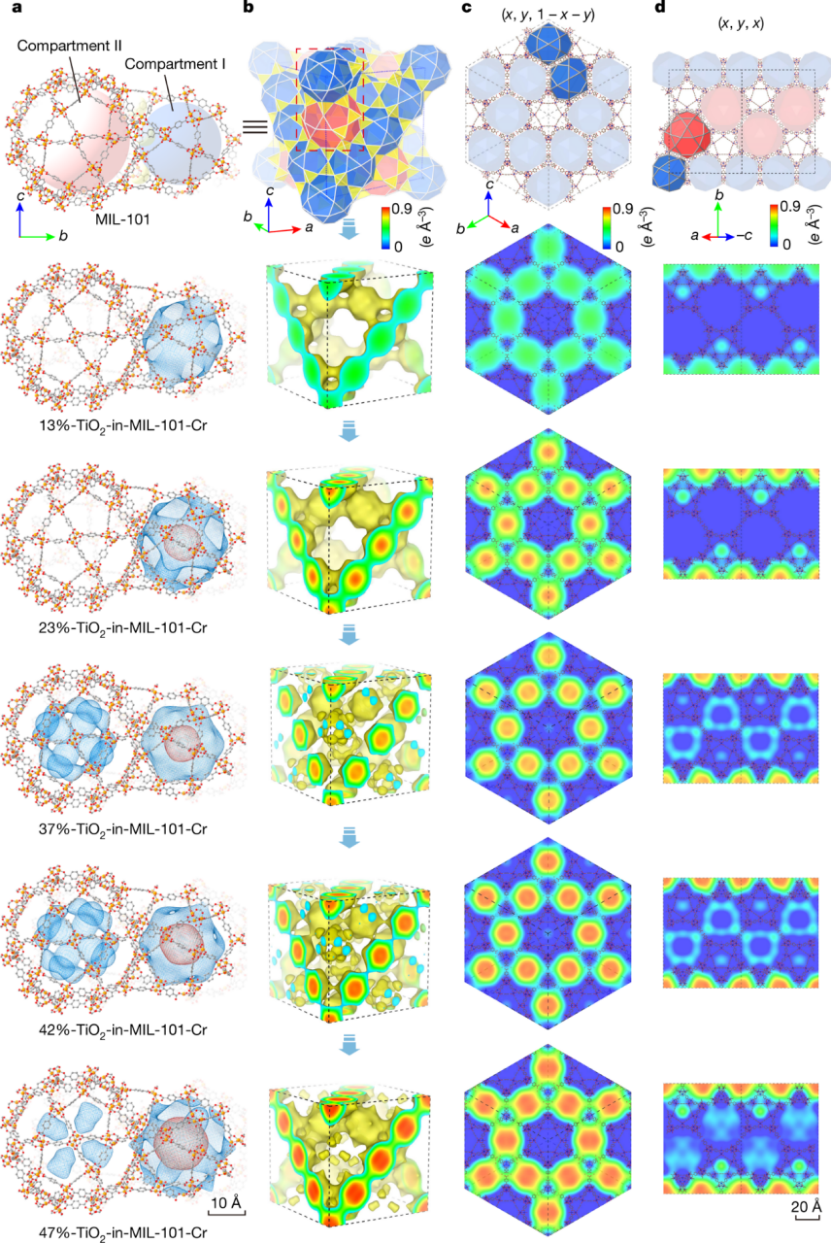
(Nature586,549–554(2020). https://doi.org/10.1038/s41586-020-2738-2)
2002年,Natasha Erdman等学者研究了SrTiO3 (001) 富TiO2表面的结构和化学性质。他们通过高分辨率电子显微镜和理论直接方法,对SrTiO3 (001) 的2×1表面结构进行解析,同时制备相关样品并利用多种电子显微镜技术进行表征。
研究发现,表面TiO6−x单元重排为边共享块决定了SrTiO3的SrO缺陷表面结构。在表面层中,存在两种不同类型的O原子,表面Ti原子通过与外部O原子更强的键合来补偿较低的配位数。理论计算部分,采用平面波赝势密度泛函(DFT)对 13 层平板模型进行计算,从实验获得的原子位置出发进行三维结构优化,确认了x,y位置并优化了z方向结构;还运用原子轨道线性组合(LCAO) DFT 计算获得部分原子电荷,为表面结构和原子间相互作用提供了有力支撑。此外,研究提出该表面结构形成规则可能适用于一般的钙钛矿表面。
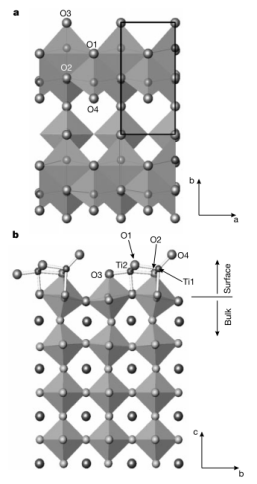
(Nature419, 55–58 (2002). https://doi.org/10.1038/nature01010)
2021年,Wentao Yuan、Beien Zhu、Ke Fang等学者利用像差校正环境透射电子显微镜(ETEM),研究了金(Au)与二氧化钛(TiO2)载体之间的界面在一氧化碳(CO)氧化反应中的变化情况。他们通过实验观察Au纳米颗粒(NPs)在TiO2表面的行为,并控制反应环境中的气体成分和温度,探究其对Au- TiO2界面结构的影响。
研究发现,在CO氧化过程中,Au NPs在TiO2(001)表面发生外延旋转,且界面结构随氧气压力和反应气体环境变化而改变。这种旋转是可逆且可控的,通过改变气体和温度可实现对活性Au- TiO2界面的原位操控。理论计算部分,采用密度泛函理论(DFT)对不同环境下Au NPs的旋转行为进行研究。构建Au116团簇模型模拟Au NPs,计算不同构型的能量、O2吸附覆盖度和电子结构。结果表明,O2吸附影响界面稳定性,CO消耗界面O2导致Au NPs旋转。同时,从电子态密度和电荷密度差计算可知,不同构型的Au NPs与 O2的结合能力不同,解释了实验中观察到的界面结构变化和催化活性差异,为理解催化反应中界面的动态变化提供了理论依据。
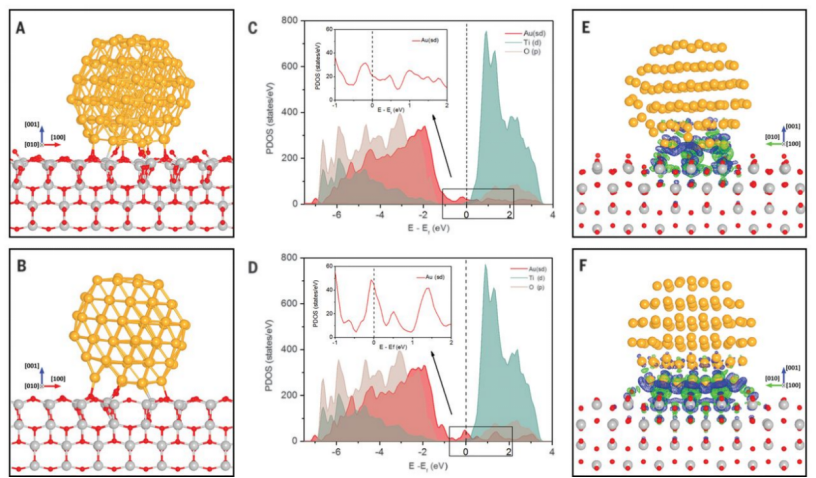
(Science 371, 517–521 (2021) 29 DOI: 10.1126/science.1207272)
Isabel Xiaoye Green、Wenjie Tang 等学者研究了Au/TiO2催化剂上 CO 氧化的反应机制。他们通过原位红外光谱技术,在低温条件下跟踪 CO 在Au和TiO2位点的吸附及反应过程中的动力学变化,并结合密度泛函理论(DFT)计算,探究该催化剂上 CO 氧化的活性位点和反应机理。
研究发现,在Au/TiO2催化剂上存在双催化位点,CO 氧化反应主要发生在TiO2位点,而非传统观点认为的仅在Au位点。在低温下,CO先从TiO2位点参与反应,且反应速率较快,而CO/Au位点的贡献较小。实验测得该反应的表观活化能为0.16±0.01eV。理论计算部分,采用梯度校正的 DFT 计算,构建了负载在金红石TiO2(110)表面的 Au 纳米棒模型来模拟实验中的纳米颗粒催化剂。计算结果表明,O2在与 Au 原子相邻的Ti5c位点吸附最强,且该位点的O2活化能最低。整个反应过程涉及O2的吸附、与CO的相互作用、O−O键的断裂、CO2的生成和扩散等步骤,各基元反应步骤的计算活化能与实验测得的表观活化能相符,为双催化位点的反应机制提供了理论支持。
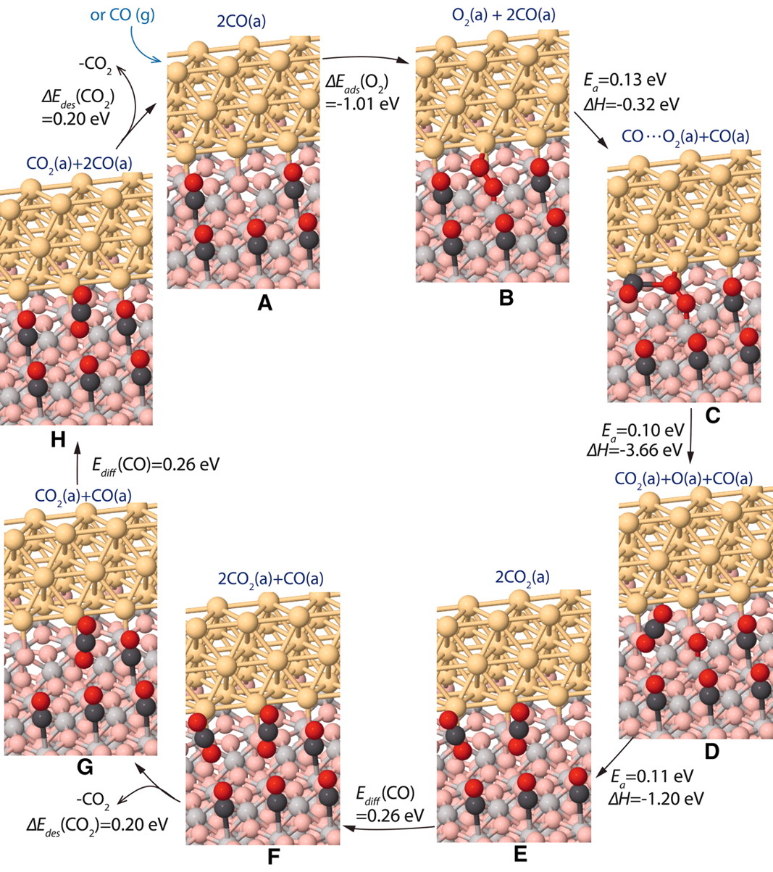
(Science333,736-739(2011).DOI:10.1126/science.1207272)
结语
TiO₂光催化技术历经50年发展,已从实验室走向实际应用。未来研究将聚焦于多尺度材料设计(如原子级掺杂、介孔结构调控)与人工智能辅助催化剂开发,以突破效率瓶颈,推动其在碳中和与环境污染治理中的大规模应用。
找华算做计算?专业靠谱省心又省时!
益于理论计算化学的快速发展,计算模拟在纳米材料研究中的运用日益广泛而深入。科研领域已经逐步形成了“精准制备-理论模拟-先进表征”的研究模式,而正是这种实验和计算模拟的联合佐证,更加增添了论文的可靠性和严谨性,往往能够得到更广泛的认可。
华算科技已向国内外1000多家高校/科研单位提供了超过50000项理论计算和测试表征服务,部分计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。
