

引言:液体电解质的挑战与AI的机遇
在锂离子电池中,液体电解质是连接正负极的“血液”,其性能直接影响电池的能量密度、循环寿命和安全性。然而,现有商用电解液多为多组分碳酸酯体系,实验优化需耗费大量时间与成本。传统分子动力学模拟虽能辅助设计,但面临两难困境:量子力学计算精度高却速度慢,经典力场速度快但精度不足。如何平衡精度与效率,成为电解质研发的核心难题。
近年来,模拟计算力场(Machine Learning Force Fields, MLFF)崭露头角。它通过拟合量子力学数据,既能保持高精度,又能大幅提升计算速度,成为分子模拟领域的“新宠”。然而,液体电解质因复杂的溶剂–离子相互作用(如溶剂分隔离子对SSIP、接触离子对CIP、聚集体AGG共存),对MLFF的泛化性和稳定性提出更高要求。
2025年4月,字节跳动研究院团队在《Nature Machine Intelligence》发表论文,提出全新框架BAMBOO(ByteDance Artificial Intelligence Molecular simulation Booster),首次将MLFF成功应用于多组分液体电解质的分子动力学模拟,并在密度、粘度、离子电导率等关键性质的预测上达到行业顶尖水平。


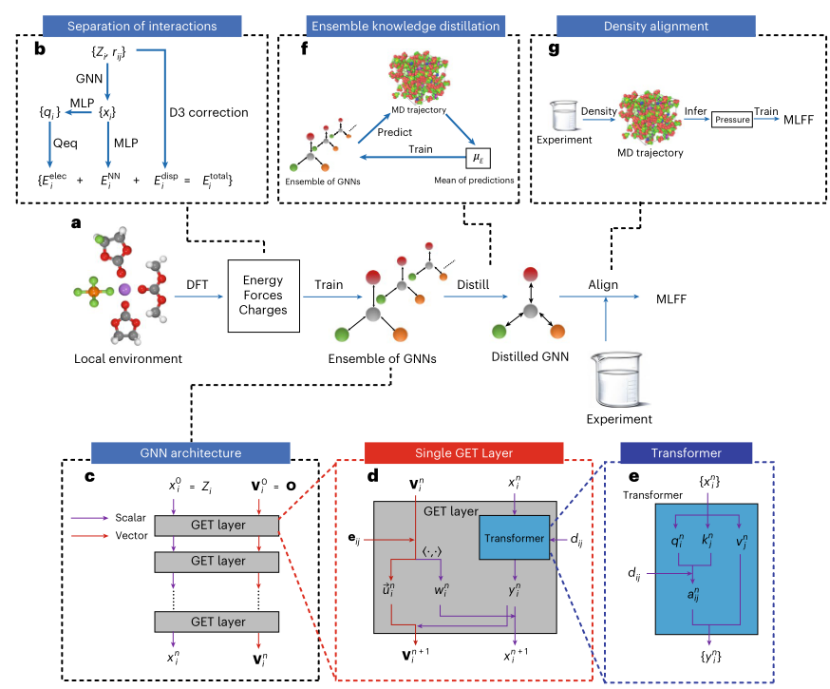
BAMBOO的核心创新:三大技术突破
1. 图等变变换器(GET):物理启发的神经网络架构
传统MLFF多基于局部描述符或普通图神经网络(GNN),难以捕捉液体电解质中长程静电作用和动态结构变化。BAMBOO的核心是一种图等变变换器(Graph Equivariant Transformer, GET),其设计灵感源于物理相互作用的分层特性:
技术细节解析:
·等变性(Equivariance):GET通过向量嵌入(vector embedding)确保模型对旋转、平移等对称性具有天然适应性。例如,无论分子如何旋转,模型对能量的预测结果保持一致,避免因坐标系变化导致的误差累积。
·注意力机制(Attention):引入Transformer模块,使模型能动态关注不同原子间的相互作用。例如,在锂离子(Li⁺)与阴离子(如PF₆⁻)的溶剂化过程中,GET能自动识别关键氧原子的电荷分布,精准捕捉离子对的动态行为。
·物理作用分离:将总能量拆分为三部分:
o半局域相互作用:由GET预测,涵盖短程键合、键角等化学键效应。
o静电作用:基于电荷平衡模型(Charge Equilibrium Model),通过多层感知机(MLP)预测原子部分电荷,再计算库仑相互作用。
o色散作用:直接采用DFT-D3色散校正,确保范德华力的准确性。
实验验证:
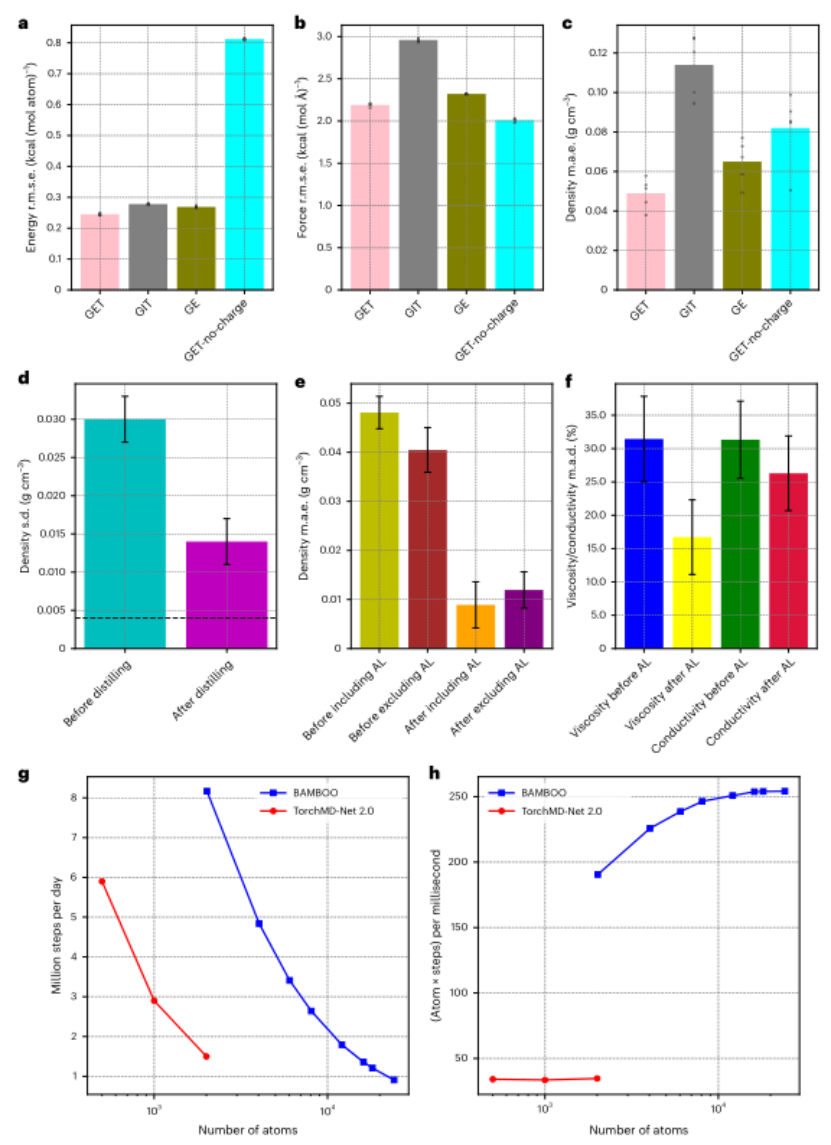
·密度预测:GET的误差(0.01 g/cm³)显著低于传统GNN模型。例如,在碳酸乙烯酯(EC)与碳酸二甲酯(DMC)的混合溶剂中,BAMBOO的预测密度与实验值几乎重合,而经典力场OPLS-AA的误差高达0.05 g/cm³。
·静电建模:显式电荷预测使离子对的动态行为更接近真实体系。例如,在高浓度LiFSI电解液中,GET成功捕捉到Li⁺电荷分布的拓宽现象(图3a-c),反映离子聚集效应。
2. 集成知识蒸馏:降低模拟波动的新思路
MLFF的随机性(如神经网络初始化差异)可能导致分子动力学(MD)模拟结果大幅波动。BAMBOO提出集成知识蒸馏(Ensemble Knowledge Distillation):
实现步骤:
1.训练多个独立模型:使用相同量子力学数据集,但不同随机种子初始化5个GNN模型。
2.生成轨迹数据:随机选择一个模型运行MD模拟,生成平衡后的分子轨迹。
3.平均预测优化:用所有模型对轨迹中的能量和力进行预测,取其平均值作为“教师信号”,微调单一模型。
效果展示:
·波动抑制:密度预测的标准差降低50%以上(从0.030 g/cm³降至0.014 g/cm³),且无需额外量子力学数据。
·通用性验证:该方法成功应用于固态相变模拟(如M3GNet模型),证明其广泛适用性。
技术意义:
集成蒸馏不仅提升了模拟稳定性,还通过“平均化”减少了模型对训练数据噪声的敏感度,尤其在处理高维、动态的液体体系时表现突出。
3. 密度对齐算法:实验与模拟的“桥梁”
量子力学数据与实验测量间常存在系统性偏差(如DFT泛函选择、团簇与体相差异)。BAMBOO通过“密度对齐(Density Alignment)”弥合这一鸿沟:
算法原理:
1.压缩率计算:通过调整模拟压力(ΔP),测量密度变化(Δρ),建立压力–密度线性关系,计算液体的压缩率(β)。
2.压力修正:利用实验密度反推所需的ΔP,通过反向传播优化MLFF参数,使模拟密度与实验值一致。
优势与效果:
·数据高效:仅需13个实验数据点,即可将密度误差从0.05 g/cm³降至0.01 g/cm³。
·迁移能力:修正效果可迁移至未参与对齐的体系。例如,对未训练的氟化溶剂(如Novec 7000),密度预测误差仍低于0.02 g/cm³。
·多性质提升:密度对齐间接优化了粘度(误差降至17%)和离子电导率(误差26%),因这些性质与分子间作用力直接相关。
物理意义:
密度对齐本质是通过宏观性质(密度)反向约束微观作用力,实现了“实验指导模拟”的闭环优化,为多尺度建模提供了新范式。
性能验证:从单一溶剂到复杂电解液
1. 基础性质预测:精度对标实验误差
·密度:在15种化学物质组成的多种体系中,平均误差仅0.01 g/cm³,与实验组内变异相当。例如,乙腈(ACT)在283K下的密度预测值为0.776 g/cm³,实验值为0.785 g/cm³,误差1.1%。
·粘度与电导率:预测偏差分别为17%和26%,显著优于经典力场OPLS-AA(图2f)。例如,对1M LiPF₆/EC电解液,BAMBOO预测粘度为4.2 mPa·s,实验值为4.0 mPa·s,而OPLS-AA预测值高达6.5 mPa·s。
·计算速度:单块NVIDIA A100 GPU可日处理200万步(10,000原子体系),效率领先同类模型。例如,BAMBOO的推理速度(6.9 ms/step)是MACE模型的2.5倍。
2. 多组分电解液:工业级复杂体系的突破
BAMBOO成功模拟了含4-8组分的实际电解液(如商用锂盐LiPF₆与碳酸酯溶剂的混合体系),精度与简单体系相当。这得益于密度对齐的强迁移性,即便训练集仅包含低浓度数据,模型仍能准确预测高浓度(3.78 mol/kg)下的性质。
案例展示:
·四元电解液(EC/DMC/EMC/LiPF₆):BAMBOO预测密度为1.25 g/cm³,实验值1.23 g/cm³,误差1.6%。而OPLS-AA的误差达8.1%。
·八元添加剂体系:加入氟代碳酸乙烯酯(FEC)、硫酸乙烯酯(DTD)等添加剂后,BAMBOO仍保持粘度预测误差低于20%。
3. 微观结构解析:从电荷分布到溶剂化工程
通过原子电荷分布的动态分析(图3),BAMBOO揭示了电解液浓度对溶剂化结构的影响:
·低浓度(1.12 m):溶剂分隔离子对(SSIP)占主导(75.1%),Li⁺电荷分布集中(峰值0.632 e)。
·高浓度(3.74 m):聚集体(AGG)占比升至54.2%,Li⁺电荷分布显著拓宽(峰值0.622 e),反映更强的离子关联。
应用价值:
此类微观洞察为“溶剂化工程”提供了新工具。例如,通过调整阴离子结构(如FSI⁻ vs. TFSI⁻),可优化Li⁺的溶剂化壳层,设计高离子迁移率的电解液。

局限性:泛化能力与未来方向
尽管BAMBOO表现出色,其泛化性仍受限于训练集的化学多样性:
·已知键型迁移:对含相似键型的新分子(如氟化溶剂),密度预测误差可控(0.05 g/cm³)。例如,氟代碳酸乙烯酯(FEC)的预测误差为0.03 g/cm³。
·未知键型挑战:如氰基(C≡N)等未训练键型可能导致模拟失稳。例如,尝试模拟含丙腈(CH₃CN)的电解液时,MD轨迹在100 ps后崩溃。
未来改进方向:
1.大规模预训练:借鉴MACE-OFF23等模型,纳入百万级分子构型数据,覆盖更广化学空间。
2.物理约束融合:将MLFF与经典力场函数结合,确保模拟稳定性,同时保留键断裂等反应建模能力。
3.多性质联合对齐:扩展密度对齐至粘度、电导率等性质的直接优化,提升综合预测能力。
产业应用:加速电池研发的“AI催化剂”
BAMBOO的落地将深刻影响电池行业:
1. 高通量筛选
传统电解液研发需合成数百种配方,耗时数年。BAMBOO可快速评估数千种溶剂/盐组合,筛选出高电导率、低粘度的候选体系。例如:
·新型锂盐设计:模拟双氟磺酰亚胺锂(LiFSI)与不同醚类溶剂的兼容性,预测其热稳定性。
·添加剂优化:筛选能抑制锂枝晶的成膜添加剂(如VC、FEC),缩短实验验证周期。
2. 微观机制指导
通过溶剂化结构与离子传输的关联分析,定向设计高性能电解液:
·低粘度电解液:增加SSIP比例,减少离子聚集(AGG),降低流动阻力。
·高低温适应性:模拟不同温度下的离子扩散行为,优化溶剂混合比例。
3. 老化与界面研究
拓展BAMBOO至电池老化模拟:
·SEI膜形成:预测电解液还原产物(如Li₂CO₃、LiF)的生成路径。
·锂枝晶抑制:分析电解液组分对锂沉积形貌的影响,指导添加剂设计。
商业化进展:
字节跳动已为BAMBOO申请中国专利(202311322469.2),并开源代码与数据集(GitHub/Zenodo),推动学术界与工业界的协同创新。特斯拉、宁德时代等企业正探索将其集成至电池研发平台。
专家观点与行业反响
·论文通讯作者Weihao Gao:“BAMBOO的核心优势在于将物理直觉与数据驱动结合,我们不仅追求预测精度,更关注模型的可解释性和工程落地。”
·斯坦福大学材料学家崔屹:“这项工作为电解液设计提供了微观尺度的‘显微镜’,未来或颠覆传统试错法研发模式。”
·宁德时代研发负责人:“我们正测试BAMBOO在新型固态电解质开发中的应用,初步结果显示模拟与实验一致性显著提升。”
未来展望:通用MLFF的星辰大海
BAMBOO的成功仅是起点,团队计划向以下方向拓展:
1.跨领域迁移:将框架应用于离子液体、聚合物电解质等体系。
2.反应动力学模拟:引入过渡态搜索算法,研究电解液分解反应路径。
3.多尺度建模:耦合宏观电池模型(如Newman模型),实现从分子到电芯的全链条优化。
读者互动:你关心的BAMBOO问题
Q:BAMBOO如何保证模拟的稳定性?
A:通过集成知识蒸馏降低随机性,同时采用物理约束(如能量守恒)避免轨迹崩溃。
Q:普通企业能否使用BAMBOO?
A:开源代码支持LAMMPS接口,企业只需具备基础算力(如A100 GPU)即可部署。
Q:BAMBOO能否替代实验?
A:目前仍是辅助工具,但可减少实验试错次数,加速研发周期。
结语:AI for Science的新里程碑
BAMBOO不仅是液体电解质模拟的工具革新,更是AI for Science范式的典型代表——通过物理启发模型架构、实验数据对齐和高效算法设计,解决传统科学计算的瓶颈问题。随着通用MLFF的发展,未来我们有望看到AI在材料、药物、能源等领域的更多突破,加速人类对复杂物质世界的理解与掌控。
论文链接:https://doi.org/10.1038/s42256-025-01009-7
代码与数据:GitHub(https://github.com/pytedance/bamboo)、Zenodo(https://doi.org/10.5281/zenodo.14603020)
找华算做计算?专业靠谱省心又省时!
益于理论计算化学的快速发展,计算模拟在纳米材料研究中的运用日益广泛而深入。科研领域已经逐步形成了“精准制备-理论模拟-先进表征”的研究模式,而正是这种实验和计算模拟的联合佐证,更加增添了论文的可靠性和严谨性,往往能够得到更广泛的认可。
华算科技已向国内外1000多家高校/科研单位提供了超过50000项理论计算和测试表征服务,部分计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。
