

VASP(Vienna Ab initio Simulation Package)是维也纳大学 Hafner 小组开发的一款进行电子结构计算和量子力学 – 分子动力学模拟的软件包,是材料模拟和计算物质科学研究中最流行的商用软件之一,以下是其详细介绍:
适用领域
1. 材料科学
新型材料研发
在寻找新型半导体、超导体、磁性材料、催化剂等方面发挥着重要作用。通过计算材料的结构、电子、光学等性质,预测新材料的性能,为实验合成提供理论指导,加速新材料的发现过程。
材料性能优化
对现有材料进行改性和优化,如研究合金化、掺杂、缺陷等对材料性能的影响。通过模拟计算,理解材料性能变化的微观机制,从而有针对性地设计材料的成分和结构,提高材料的性能。
2. 凝聚态物理
电子结构研究
用于研究各种凝聚态物质的电子结构,如金属、半导体、绝缘体等的能带结构、态密度等。通过对电子结构的精确计算,深入理解材料的物理性质,如导电性、磁性、光学性质等的微观起源。
强关联体系研究
在研究具有强电子 – 电子相互作用的体系,如高温超导体、莫特绝缘体等方面也有重要应用。VASP 软件能够采用适当的方法处理强关联效应,为揭示这些复杂体系的物理机制提供理论支持。
3. 表面科学与催化
表面结构与性质
研究材料表面的结构、电子性质以及表面吸附行为。通过计算表面能、吸附能等参数,了解物质在材料表面的吸附和反应机理,为表面工程、腐蚀防护等领域提供理论依据。
催化反应机理
在催化领域,VASP 可用于研究催化剂表面的活性位点、反应物的吸附和活化过程以及催化反应的中间步骤和机理。帮助设计高效的催化剂,提高催化反应的效率和选择性。
4. 能源领域
电池材料
对电池电极材料的结构和性能进行研究,如锂离子电池、钠离子电池等电极材料的嵌锂 / 钠过程、电子结构变化、体积膨胀等。通过计算优化电极材料的结构,提高电池的能量密度、循环稳定性和充放电效率。
光电器件
研究太阳能电池材料的光学和电子性质,如半导体光吸收材料的能带结构、激子行为等。为设计高效的光吸收层、电荷传输层等提供理论指导,提高光电器件的光电转换效率。
5. 纳米技术
纳米材料性能
研究纳米材料的独特物理化学性质,如纳米颗粒、纳米管、纳米薄膜等的尺寸效应、表面效应、量子限域效应等。通过计算模拟,揭示纳米材料性能与结构之间的关系,为纳米材料的合成和应用提供理论支持。
纳米器件设计
在纳米电子器件、纳米传感器等领域,VASP 可用于模拟器件的电子输运性质、电荷转移过程等。帮助优化纳米器件的结构和性能,推动纳米技术的发展和应用。
安装与部署
安装前准备
确保系统具备基本的编译工具和相关库。在 Linux 系统中,可使用以下命令安装必要的软件包:sudo apt – get install make build – essential g++ gfortransudo apt – get install libblas – dev liblapack – dev libopenmpi – dev libscalapack – mpi – dev libfftw3 – dev
获取安装包
从 VASP 官方网站或授权渠道获取 VASP 安装包,通常为.tar.gz格式的压缩文件。
解压与修补
打开安装包所在文件夹,执行以下命令进行解压和修补:tar – zxvf vasp.5.4.4.tar.gzgunzip patch.5.4.4.16052018.gzcd vasp.5.4.4patch – p0
配置 Makefile
将arch文件夹中的makefile.include.linux_intel拷到上一级目录下改名为makefile.include。打开此文件,根据需要修改相关参数,例如可以在OFLAG参数里加入-xhost。
编译
在修改完makefile.include后,执行以下命令进行编译,构建 VASP 的std、gam和ncl:make std gam ncl编译过程可能会持续一段时间,根据系统性能和安装包大小而异。
设置环境变量
编译完成后,在bin文件夹中会出现可执行文件。为了能够在任意目录下方便地运行 VASP,需要将 VASP 的bin目录添加到系统环境变量中。
例如,在~/.bashrc文件中添加:export PATH=”$PATH:/home/xxxx/vasp.5.4.4/bin”将/home/xxxx/vasp.5.4.4/bin替换为实际的 VASP 安装路径。保存.bashrc文件并更新环境变量,可执行source ~/.bashrc使设置生效。
理论基础
● 密度泛函理论(DFT)
基本原理
DFT 是 VASP 软件的核心理论基础。它指出体系的基态能量是电子密度的泛函,通过求解 Kohn – Sham 方程来确定电子密度和体系能量。与传统的多体理论相比,DFT 将多电子问题转化为单电子问题,大大降低了计算量,使得对较大体系的计算成为可能。
交换关联泛函
在 DFT 中,交换关联能是电子密度的泛函,但其具体形式未知,需要通过近似来处理。常见的交换关联泛函有局域密度近似(LDA)和广义梯度近似(GGA)等。LDA 假设电子密度在局域上是均匀的,而 GGA 则考虑了电子密度的梯度信息,通常能更准确地描述材料的性质。
● 平面波基组与赝势
平面波基组
VASP 采用平面波作为基函数来展开电子波函数。平面波具有完备性和正交性,能够精确地描述晶体中的电子态。通过将电子波函数表示为平面波的线性组合,可以方便地进行数值计算。
赝势
由于原子核与电子之间的相互作用非常复杂,直接计算会带来很大的计算量。赝势方法将核心电子与价电子分开处理,用一个赝势来代替原子核和核心电子对价电子的作用。这样可以有效地减少计算量,同时保持计算结果的准确性。
运行流程
1. 输入文件准备
结构文件
通常使用 POSCAR 文件来描述晶体的结构信息,包括晶格参数、原子种类和坐标等。可以通过晶体结构数据库或实验数据获取初始结构信息,也可以根据研究目的自行构建结构模型。
计算参数文件
INCAR 文件用于设置各种计算参数,如平面波截断能量、K 点网格、交换关联泛函、计算精度、弛豫算法等。这些参数的设置会直接影响计算的结果和效率,需要根据具体的研究体系和要求进行合理调整。
赝势文件
VASP 需要使用赝势文件来描述原子的赝势信息。不同的原子种类有对应的赝势文件,用户需要根据体系中的原子选择合适的赝势,并将其路径正确设置在输入文件中。
2. 结构弛豫
初始结构优化
VASP 首先会读取输入文件中的初始结构信息,并根据设置的计算参数进行结构弛豫。在这个过程中,软件会通过计算原子所受的力,采用一定的优化算法(如共轭梯度法、BFGS 法等)来调整原子的位置,使体系的总能量达到最小,得到稳定的结构。
晶格参数优化
除了原子位置的优化,VASP 还可以对晶格参数进行优化。通过改变晶格参数,使体系的能量在晶格空间中达到最小值,从而得到优化后的晶格结构。结构弛豫的收敛标准通常以原子受力和能量变化小于一定的阈值来判断。
3. 自洽计算
电子结构计算
在优化后的结构基础上,VASP 进行自洽场(SCF)计算,以确定材料的电子结构。首先,软件会根据输入参数初始化电子密度,然后通过迭代求解 Kohn – Sham 方程,逐步更新电子密度和能量,直到电子密度和能量的变化满足自洽收敛条件。
输出结果
自洽计算收敛后,VASP 会输出一系列结果文件,包括总能量、电子态密度、能带结构、电荷密度等信息。这些结果可以用于分析材料的各种物理性质,如导电性、磁性、光学性质等。
4. 性质计算
根据需求选择计算
在得到电子结构的基础上,用户可以根据研究目的选择计算材料的其他性质,如光学性质、力学性质、磁学性质等。这些性质的计算通常需要调用相应的模块和算法,结合电子结构的结果进行进一步的计算和分析。
结果分析与讨论
对计算得到的各种性质结果进行分析和讨论,与实验数据或其他理论计算结果进行对比,以验证计算的准确性和可靠性。同时,通过对计算结果的深入分析,揭示材料的微观结构与宏观性质之间的关系,为材料的设计和优化提供理论依据。
5. 后处理
数据可视化
为了更直观地理解计算结果,通常需要使用可视化软件对输出的数据进行处理和展示。例如,可以使用 VESTA、XCrySDen 等软件来可视化晶体结构、电荷密度、能带结构等信息,帮助用户更好地分析和解释计算结果。
结果总结与报告
将计算得到的重要结果进行总结和整理,撰写研究报告或论文。在报告中,需要详细描述计算方法、模型设置、主要结果以及对结果的分析和讨论,以便与同行进行交流和分享研究成果。
计算内容
1. 结构性质
晶格参数优化
确定材料的稳定晶格结构,计算出晶格常数、晶胞体积等参数,使晶胞的能量处于最低状态。
原子位置弛豫
找到原子在晶格中的最优位置,得到体系的稳定结构,可用于研究材料在不同条件下的结构变化。
晶体结构预测
通过计算不同结构的能量,预测材料可能存在的稳定晶体结构,为新材料的发现提供理论指导。
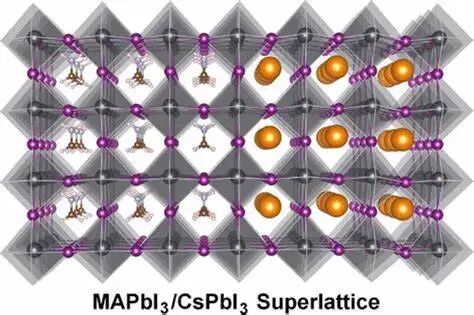

2. 电子性质
能带结构
计算电子在晶体中的能量分布,得到能带结构图像,从而分析材料的导电性、半导体特性、金属性等,确定材料是导体、半导体还是绝缘体,以及能隙的大小。
态密度
包括总态密度(TDOS)和分波态密度(PDOS),能了解电子在不同能量区间的分布情况,以及不同原子轨道对电子态的贡献,分析材料的成键特性和电子结构。
电荷密度
直观地展示电子在空间中的分布情况,通过分析电荷密度分布,可以了解原子之间的成键方式、化学键的性质以及电荷转移情况。
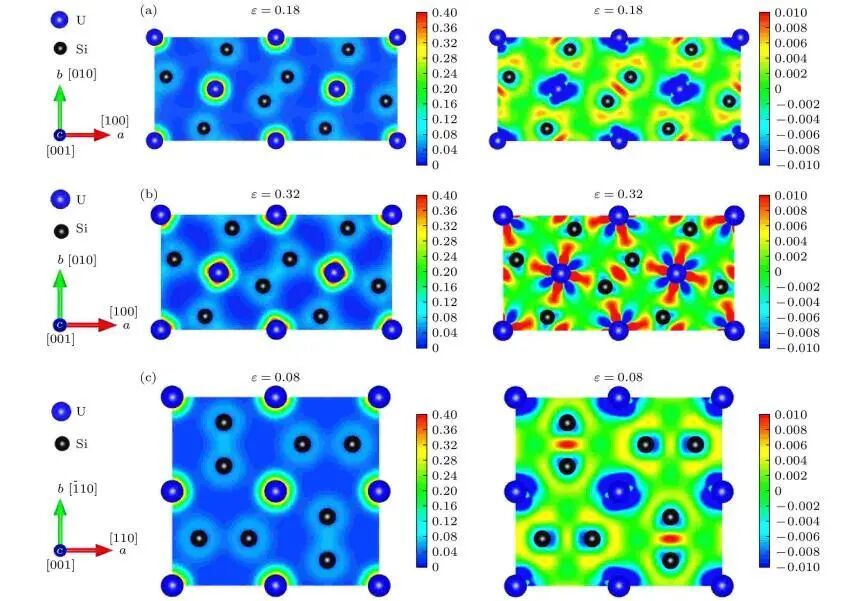
3. 光学性质
介电函数
计算材料的介电函数,进而得到反射率、吸收率、折射率等光学常数,以此研究材料对不同频率光的响应特性,为光电器件的设计提供理论依据。
光学吸收光谱
理论模拟材料的光学吸收谱,了解材料在不同波长光下的吸收情况,可用于分析材料的光吸收性能、光电转换效率等。
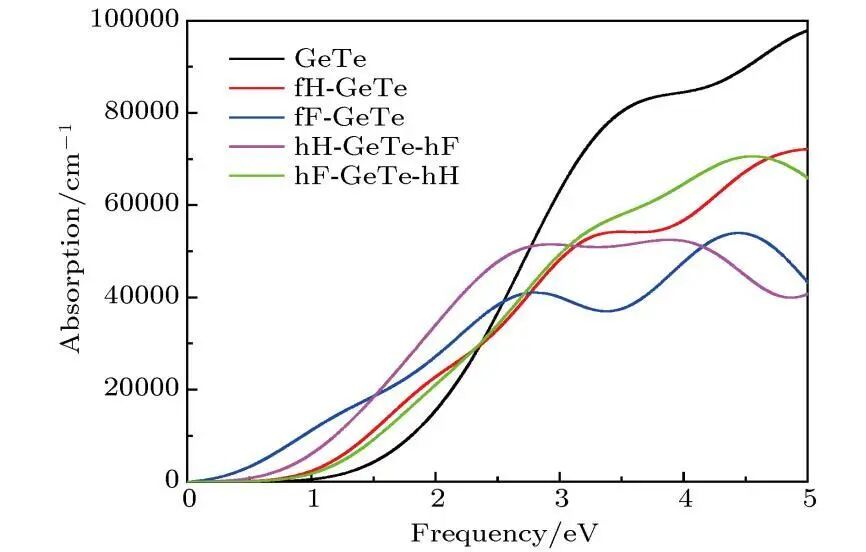
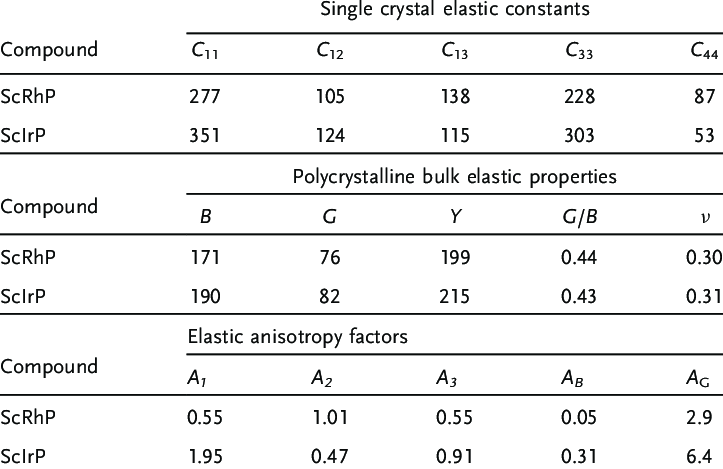
4. 力学性质
弹性常数
计算材料的弹性常数,如杨氏模量、剪切模量、体积模量等,以评估材料的刚度、韧性和稳定性,为材料在机械工程等领域的应用提供力学性能方面的参考。
硬度
通过理论计算预测材料的硬度,有助于筛选适合特定应用场景的高硬度材料或设计具有良好力学性能的复合材料。
5. 磁学性质
磁矩
计算材料的总磁矩以及原子磁矩,研究材料的磁性来源和磁性大小,判断材料是铁磁性、反铁磁性还是顺磁性等。
磁交换相互作用
分析磁性原子之间的磁交换相互作用,了解材料磁性的微观机制,对于设计和开发具有特定磁性性能的材料具有重要意义。
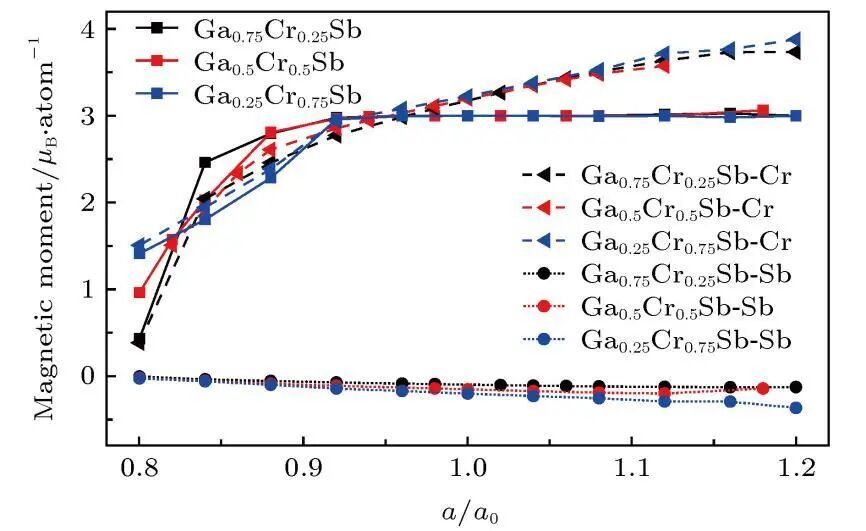
6. 热学性质
比热容
计算材料的比热容,了解材料在不同温度下吸收和储存热量的能力,为材料在热管理、能源存储等领域的应用提供热学性能参数。
热膨胀系数
理论预测材料的热膨胀系数,研究材料在温度变化时的尺寸变化规律,对于高温材料的设计和应用具有重要指导作用。
本页内容为VASP零基础系统化教程的单个章节。为了让您的学习更加高效、体系化,避免碎片化知识的困扰,建立坚实的知识框架,我们诚邀您访问 【VASP零基础系统化教程合集】 页面。该合集整合了全部教程,形成了一套严谨的 “理论奠基 → 工具准备 → 参数实操 → 场景实战 → 结果分析” 四阶一闭环学习体系。遵循此路径,您将能循序渐进地掌握VASP计算模拟的核心技能。
【做计算 找华算】
? 华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服务。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???