

Q1:老师我减去氢气的CHGCAR的时候会有这种报错(三维数据不匹配?)然后不能进行差分,这应该怎么处理啊。
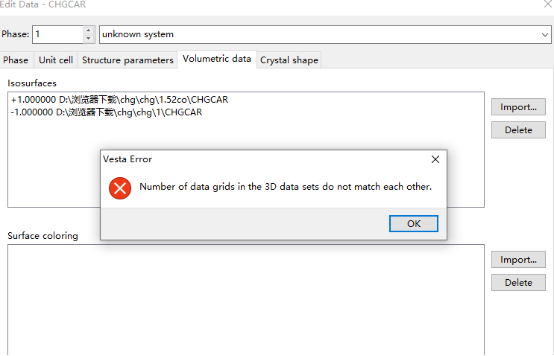

A:检查一下三个计算的ngxf是否一样,不一样就会出错
Q2:朱老师,bader分析中出现这个问题是什么原因?
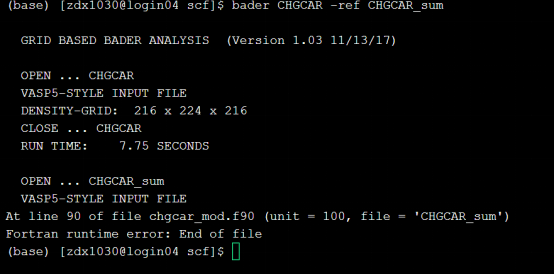
A:之前chgsum.pl有没有运行
Q3:朱老师,体系中有Mn元素,赝势用Mn还是Mn_pv呢?
A:首选Mn
Q4:朱老师,显式溶剂模型中如果想模拟碱性环境,应该怎么根据pH确定碱金属原子的数目呢?
A:只能象征性放一些
Q5:朱老师,为什么会出现红框里这些东西,应该怎么解决?
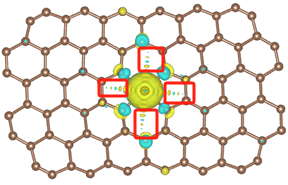
A:把等值面的值调大一些
Q6:朱老师,结构优化和电子结构计算k点不一样,和两个设置一样对比结果影响大吗?

A:优化可以少点,后面多点,没关系
Q7:老师,在算掺杂时,如何选取掺杂位点呢,最后是要选取掺杂后结构优化的能量最低那个吗?
A:对的,能量最低。
Q8:朱老师,之前咨询过您这个问题,就是NaYF4算表面能relax模型放开表层原子时ALGO无论是用N,F还是V,scf都无法收敛,您之前建议我加上AMIX参数,我设置了AMIX=0.2或者0.02都依然无法收敛,这时我还应该怎么办呢?
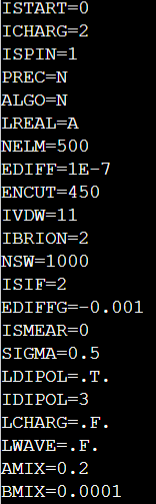
A:打开自旋试试
Q9:朱老师,NaYF4在实验中没有磁性,这时候还要打开自旋吗?我不知道磁矩该怎么设置。
还有一点,在同一个模型,如果我没有放开表面原子算能量,scf是可以收敛的。但是如果我放开表层原子,再加上偶极修正IDIPOL=3, scf就完全无法收敛了
A:y元素还是考虑自旋比较好,不收敛可以调节amix。
Q10:朱老师,氢气解离终态计算某一个氢可以固定吗,上面第一张是我氢气全放开算的,吸附在氮上的H偏向Co,下面是文献里的就只吸附在氮的正上方,为什么会这样啊?
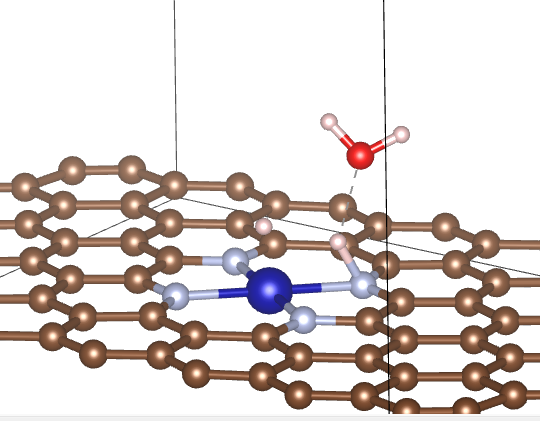
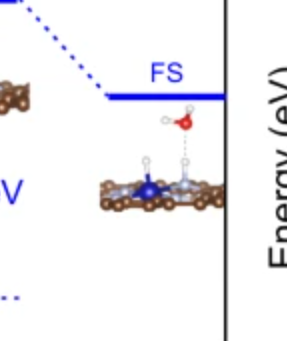
A:一般不能固定,需要找到两个吸附位点
本次答疑由拥有15年VASP实战经验的华算科技朱老师(同济大学本博、深圳海外高层次人才)提供。?点击进入《VASP | 理论计算常见问题解答专题》,快速查找更多计算解决方案。