

说明:本文华算科技从理论计算的角度,系统介绍声子(Phonons)和晶格振动(Lattice Vibrations)的基本概念、核心原理及其在固体物理中的研究进展。
内容涵盖声子和晶格振动的定义、热力学特性、主要计算方法(如密度泛函理论)以及在热传输、超导和材料设计中的重要性。
读者可通过本文了解声子和晶格振动的量子机制、模拟技术的关键作用,以及其在先进材料系统设计中的潜力,为计算物理、材料科学和凝聚态物理的创新研究提供理论支持和实践指导。
什么是声子和晶格振动?
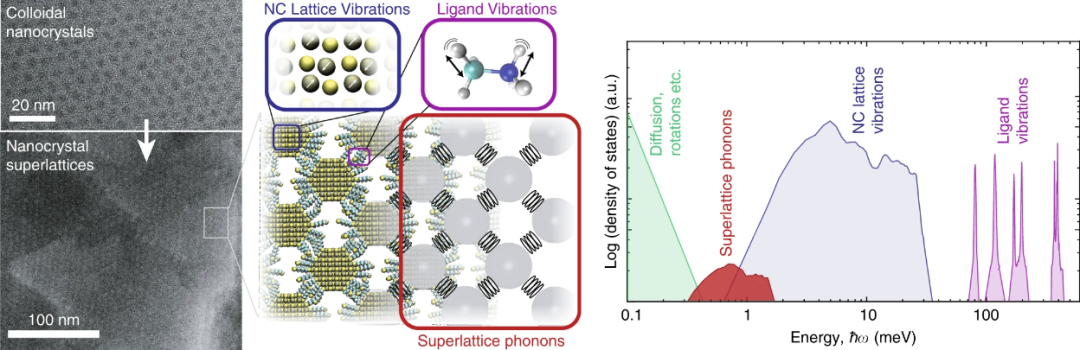

DOI: 10.1038/s41467-019-12305-3
声子和晶格振动是指固体晶体中原子集体振动的量子化描述,声子作为准粒子表征这些振动模式,直接影响材料的热、电和光学性质。
晶格振动分为声学和光学分支,由原子间谐振相互作用驱动;在金属或半导体中,声子通过与电子耦合影响超导和热导率。
其核心原理源于Born-von Karman模型和量子谐振子理论,振动频率由晶格对称性和力常数决定,调控热振动势垒以影响材料稳定性。
传统实验方法如中子散射可表征振动谱,但理论计算方法在揭示电子–声子耦合和动态响应方面具有独特优势。这些计算工具不仅能预测声子色散关系,还能评估其对热传输和相变的影响,推动从原子尺度到器件应用的材料设计。
声子和晶格振动的理论计算方法
理论计算在声子和晶格振动研究中扮演关键角色,用于预测振动谱、耦合机制和性能优化。以下介绍主要计算方法及其在声子和晶格振动中的应用。
密度泛函理论(DFT)
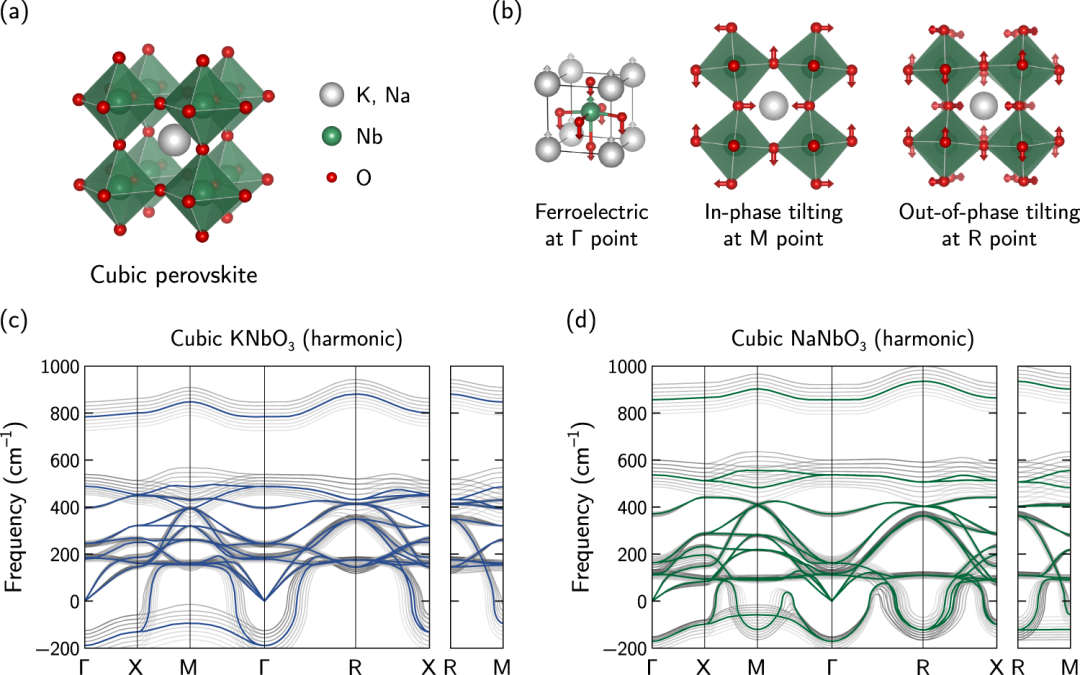
DOI: 10.1038/s41524-023-01110-8
密度泛函理论基于量子力学,计算晶格振动的电子结构、力常数和声子频率,是研究声子和晶格振动最常用的方法。其核心优势是无需经验参数,直接从电子密度层面预测原子间相互作用和振动模式。
例如,采用密度泛函理论(DFT)和自洽声子(SCP)计算来研究立方铌酸盐钙钛矿中的有限温度声子。
为了包含明确的非简谐振动效应,SCP 频率通过准粒子(QP)近似内的气泡自能校正进行移动,从而提供了这些强非简声固体中声子软化的精确描述。
其他方法
其他方法包括分子动力学模拟(MD)和机器学习(ML),为特定场景提供补充。
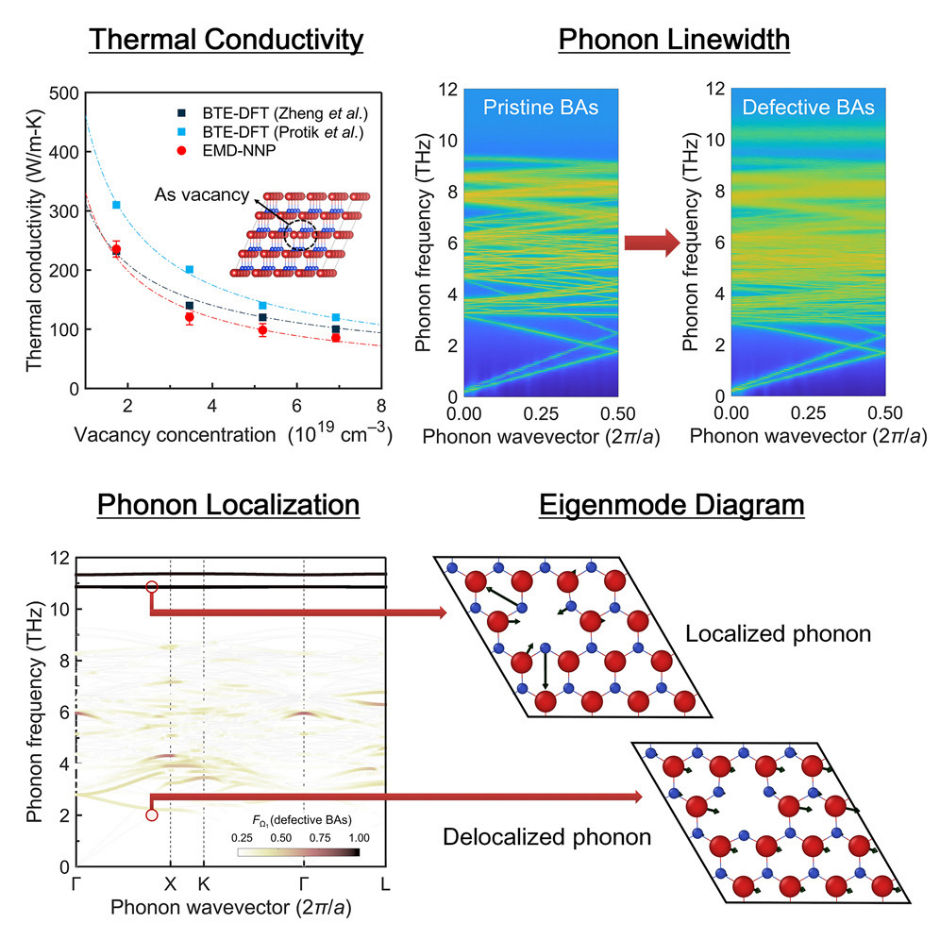
DOI: 10.1016/j.xcrp.2023.101760
MD模拟:通过微扰响应直接计算声子频率,避免有限位移方法的数值噪声,适合高通量筛选。
例如,通过分子动力学模拟,我们定量探讨了砷空位引起的砷化硼中声子局域化的程度,强调了分子动力学模拟与神经网络原子间势相结合对缺陷系统的适用性,为实际半导体晶体中的声子工程奠定了理论基础。
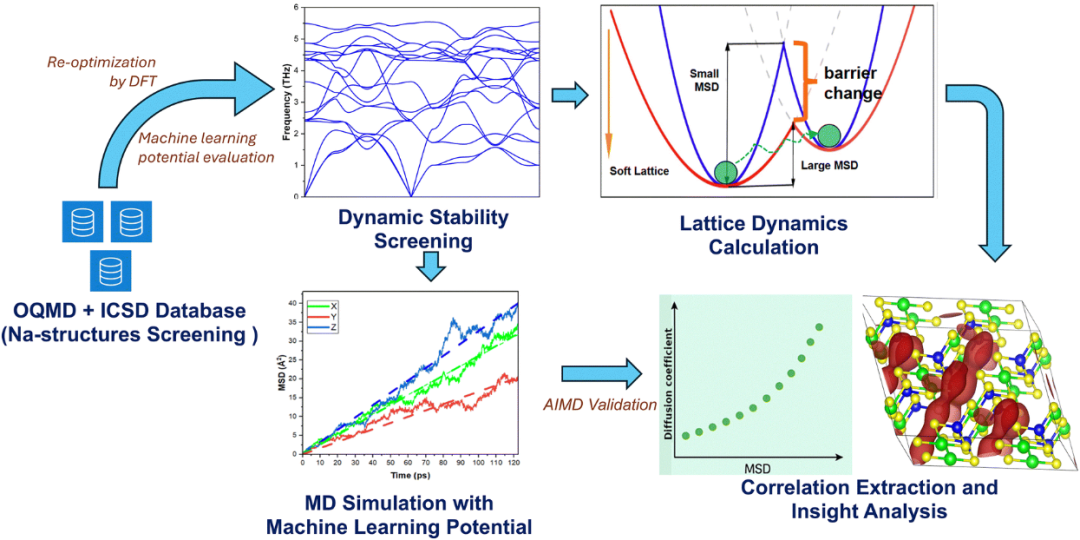
DOI: 10.1039/d5mh01176k
机器学习:基于经典力场模拟非谐振动和有限温度效应,适用于大尺度晶体动态分析。例如,通过分析Na+离子的声子均方位移(MSD)确定了控制离子电导率的关键晶格动力学特征。
通过对 3903 个含钠结构的数据集进行高通量筛选,我们建立了声子 MSD 与扩散系数之间的强正相关关系,提供了晶格动力学和离子传输之间的定量相关性。
结论
声子和晶格振动作为固体材料动态的核心,通过精确的量子模式和耦合机制实现热电调控,成为凝聚态物理和材料工程的焦点。
密度泛函理论作为主要计算工具,通过电子结构建模和声子色散分析,为机制解析和优化提供了强大支持。
其他方法如MD和机器学习进一步扩展了应用场景,显著推进了热传输、超导和可持续材料中的研究。
随着计算技术和算法的进步,如高通量DFPT的集成,声子和晶格振动的设计将进一步加速,为先进能源和电子器件提供新机遇。
【做计算 找华算】
? 华算科技提供专业的第一性原理、分子动力学、生物模拟、量子化学、机器学习、有限元仿真等代算服务。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???