VASP(Vienna ab initio simulation package)是一种广泛应用于材料科学和化学计算的软件包,尤其在第一性原理计算中具有重要地位。
VASP基于密度泛函理论(DFT)进行电子结构计算,能够模拟材料的电子结构、动力学行为、热力学性质等。
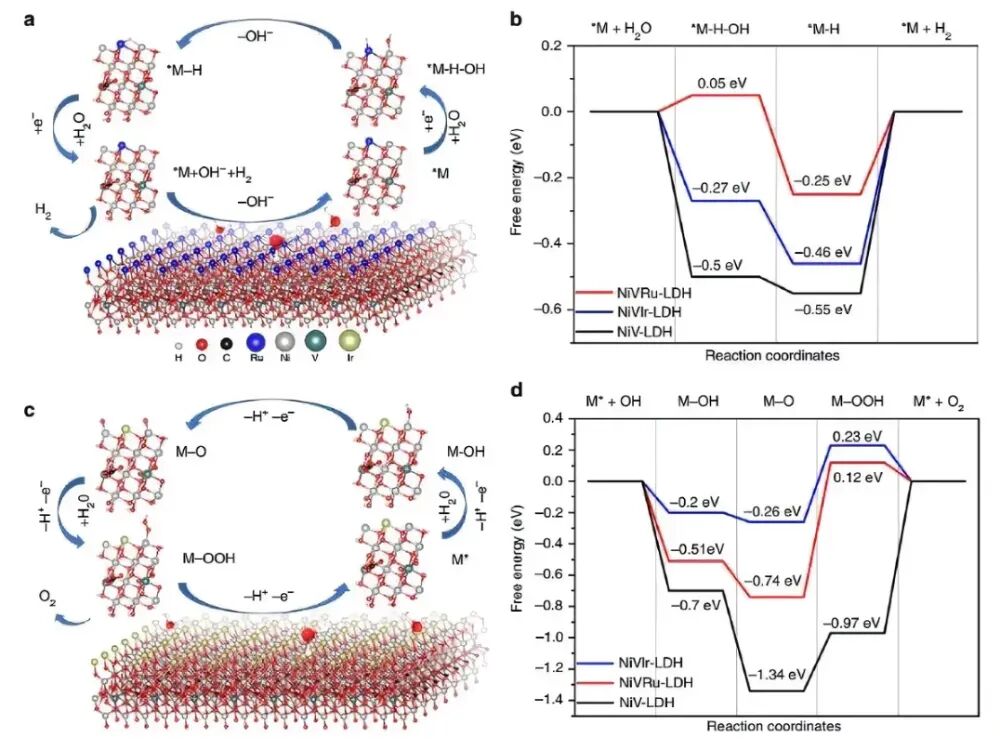
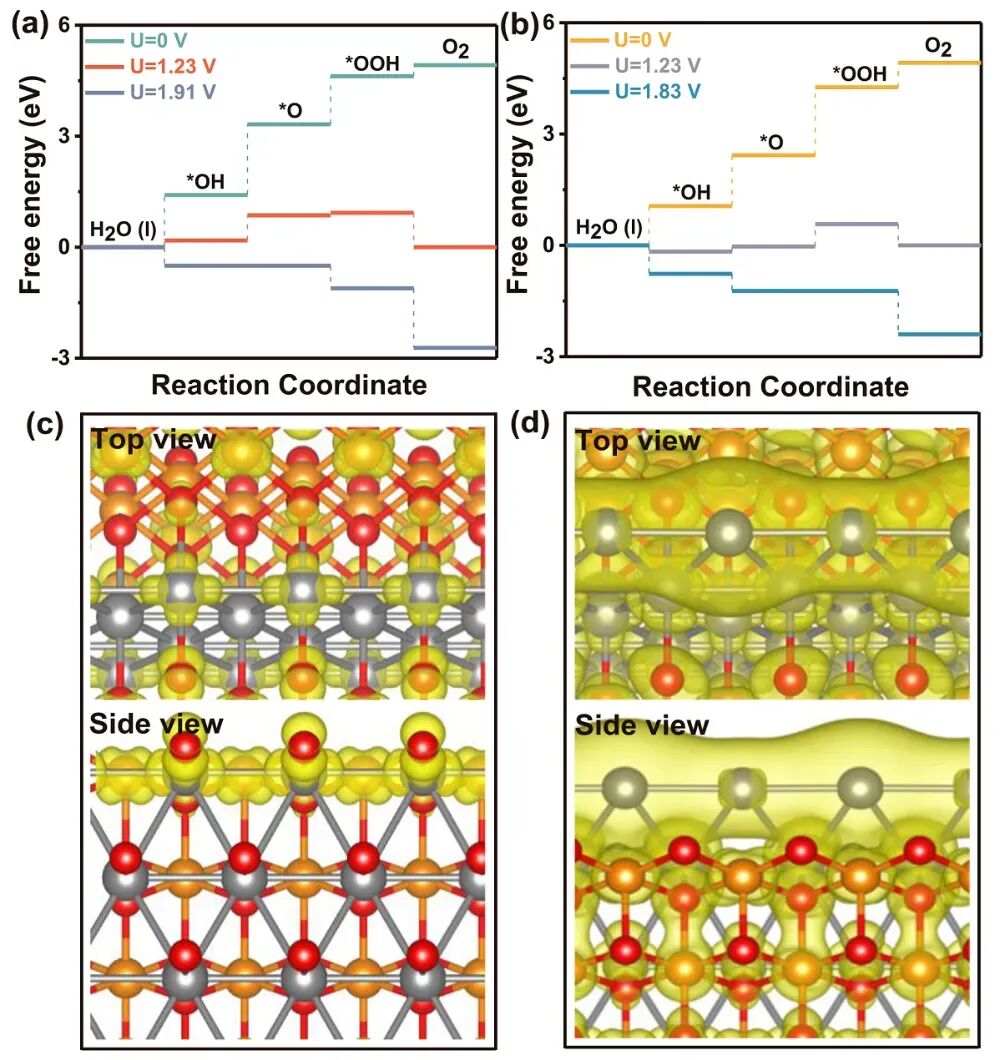
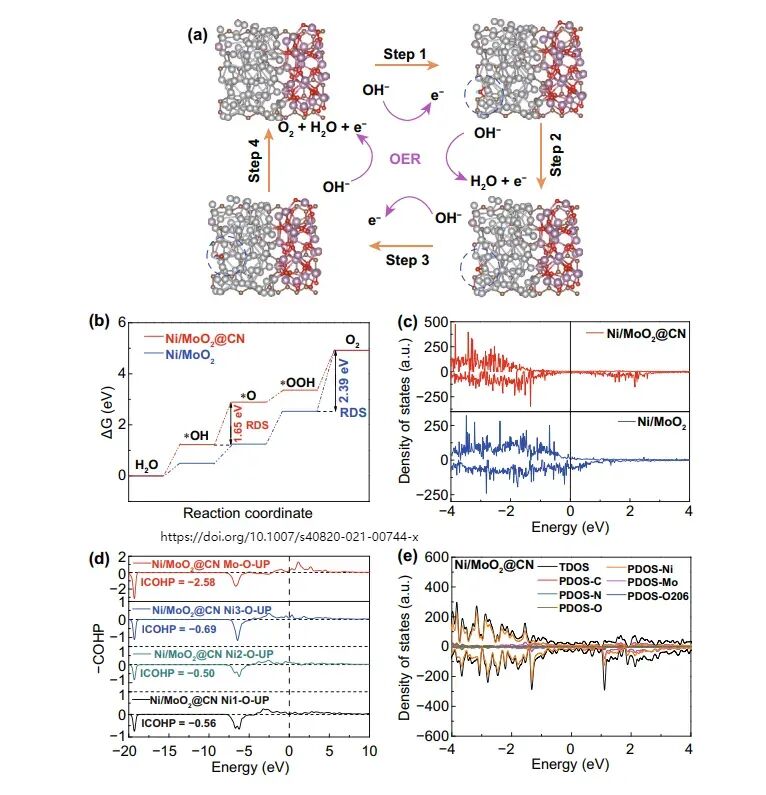
华算科技朱老师发现在电催化领域,VASP被广泛用于计算氧析出反应(OER)的自由能台阶图,以分析反应路径和优化催化剂性能。
VASP的核心在于其高效的平面波基组和赝势方法,能够高效地计算大系统的电子结构。VASP的计算流程包括结构建模、参数设置、计算收敛和结果分析等步骤。在电催化计算中,VASP常用于计算吸附能、自由能变化和反应路径。
氧析出反应(OER)是一个多步反应过程,涉及水分子的吸附、中间体的形成和产物的释放。
自由能台阶图是分析OER反应路径的重要工具,通过绘制反应路径中各步骤的能量变化,可以识别决速步骤(RDS)并优化催化剂性能。
搭建用于计算的结构模型,包括吸附模型(如H₂O、OH*、O*、OOH*、O₂等)。
设置INCAR、KPOINTS和POSCAR文件,包括计算参数(如ISIF、ISPIN、EDIFF、EDIFFG等)。
将各步骤的自由能变化绘制成台阶图,分析反应路径和催化剂性能。
在VASP计算中,优化计算效率和精度是关键。以下是一些常用的技巧
调整NCORE、KPAR、ENCUT等参数以提高计算效率。
设置合适的EDIFF、EDIFFG等收敛标准,确保计算结果的准确性。
使用CI-NEB方法进行过渡态计算,以寻找反应路径中的关键点。
在VASP计算完成后,通过分析OUTCAR、DOSCAR等输出文件,提取计算结果。对于OER台阶图,通常需要将各步骤的自由能变化绘制成图,以直观展示反应路径和催化剂性能。
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!