

说明:本文华算科技系统介绍了自组装分子动力学模拟的原理、标准流程(构建–最小化–平衡–生产–分析)、关键应用(如软物质与生物大分子组装)及核心技术挑战(如尺度与力场问题)。
通过阅读,您将掌握这一在原子尺度揭示分子自发有序组装过程的强大工具,学会如何利用它来研究材料形成机制、药物聚集行为或生命过程,为您在化学、材料和生物领域的科研工作提供关键的模拟方法与分析思路。
什么是自组装分子动力学模拟


要理解自组装分子动力学模拟,首先需要把握其两个核心组成部分:分子动力学(Molecular Dynamics, MD)与自组装过程。
分子动力学是一种计算机模拟方法,其理论基础是经典牛顿力学。它将系统中的每个原子视为一个经典粒子,通过求解牛顿运动方程来计算在给定时间步长内系统中每个原子的受力、速度和位置变化,从而描绘出整个分子体系随时间演化的动态轨迹。
这一过程的核心在于“力场”(Force Field),它是一套复杂的数学函数和参数,用以精确描述原子间的相互作用,包括维系分子结构的键合作用(如键长、键角、二面角)和决定分子间如何聚集的非键合作用(如范德华力、静电相互作用)。
自组装分子动力学模拟本身并非一种全新的、独立的模拟理论,而是分子动力学方法在研究自组装现象这一特定问题上的应用。它与常规的MD模拟在底层物理原理和算法上是完全一致的,其根本区别在于研究的目标和初始设置。
常规MD模拟通常从一个已知的、相对稳定的结构(例如从蛋白质数据库PDB中获得的蛋白质晶体结构)出发,研究其在特定环境下的局部构象变化、稳定性或与其它分子的相互作用。
而自组装模拟的目标是观察从无序到有序的完整过程,因此其初始状态通常是将大量分子单元以随机、无序的方式分布在模拟盒子中,并设定特定的浓度和溶剂环境。
通过长时间的模拟,研究人员可以像观看一部“分子电影”一样,实时追踪这些独立的分子单元如何通过相互识别、碰撞、结合,最终自发形成如胶束、囊泡、纤维、晶体等复杂的超分子结构。
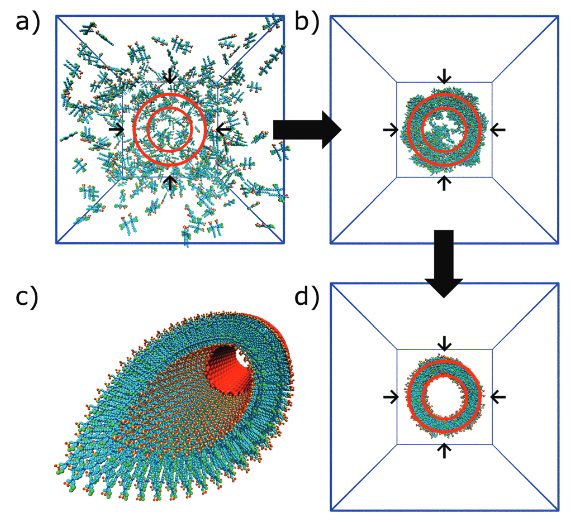
图1分子自组装成双层管状结构,DOI: 10.1039/c8cs00040a
自组装分子动力学模拟的流程
自组装分子动力学模拟流程可大致分为体系构建、能量最小化、系统平衡、生产运行和数据分析五个核心阶段,每一步都对最终结果的准确性和可靠性至关重要。
1.体系构建(System Building)
这是自组装模拟最具特色的步骤。首先,需要构建单个分子单元的结构,这可以通过化学绘图软件完成,并利用合适的力场(如CHARMM, AMBER, OPLS等)为其生成拓扑和参数文件。
随后,使用GROMACS、LAMMPS等模拟软件包将预设数量(决定了体系浓度)的分子单元随机地、或以网格状地填充到一个设定好尺寸的模拟盒子中。
最后,向盒子中填充溶剂分子(如水),并加入适量抗衡离子以中和体系电荷,确保模拟环境的真实性。这一步的关键在于确保初始构象的随机性,从而让体系能够自由探索其自组装路径。
2.能量最小化(Energy Minimization)
由于初始的随机放置,分子间可能存在不合理的空间重叠或过近的距离,导致极高的初始势能和作用力,这会使模拟直接崩溃。能量最小化步骤通过最陡下降法或共轭梯度法等算法调整原子坐标,消除这些不合理的构象,使体系达到一个临近的局部能量最低点,为后续的动力学模拟准备一个稳定的出发点。
3.系统平衡(Equilibration)
经过能量最小化的体系仍处于非平衡态。为了使其达到预设的实验条件(如特定温度和压力),需要进行系统平衡。这个过程通常分为两个阶段:首先是NVT系综(粒子数N、体积V、温度T恒定)平衡,通过温度耦合算法(如V-rescale或Nosé-Hoover)将体系加热至目标温度并使其稳定。
随后是NPT系综(粒子数N、压力P、温度T恒定)平衡,在维持温度的同时,引入压力耦合算法(如Parrinello-Rahman或Berendsen)调整盒子体积,使体系密度和压力也达到并稳定在目标值。
4.生产运行(Production Run)
当体系的温度、压力、密度等宏观参数在平衡阶段的末期不再有大的波动时,就可以撤去所有限制,开始正式的生产运行。这是模拟的核心阶段,其目的是在设定的系综下(通常是NPT)进行尽可能长时间的模拟,以捕捉到缓慢的自组装过程。
模拟的时长是自组装模拟成功的关键,往往需要从几十纳秒到数微秒甚至更长。在这一阶段,系统会以固定的时间间隔(例如每10皮秒)保存所有原子的坐标信息,生成轨迹文件(trajectory file),这是后续分析的数据基础。
5.数据分析(Analysis)
生产运行结束后,研究人员将利用轨迹文件进行深入的后处理分析。针对自组装研究,常见的分析包括:计算团簇(cluster)尺寸随时间的变化来研究组装动力学;计算径向分布函数(RDF)来表征组装体的结构有序性;计算均方根偏差(RMSD)来评估组装体的稳定性;通过可视化软件(如VMD、PyMOL)直观地观察组装形态;计算溶剂可及表面积(SASA)来分析疏水作用的贡献;以及利用高级采样方法计算自组装过程的自由能变化等。
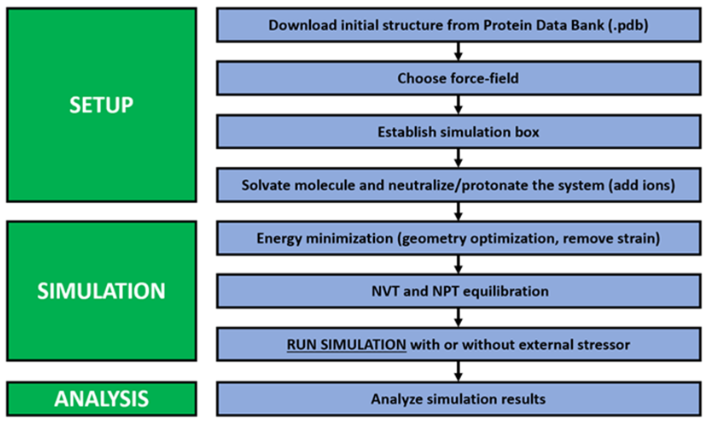
图2使用Gromacs运行自组装模拟的一般步骤
自组装分子动力学模拟的应用领域
凭借其在原子尺度上提供时空连续动态信息的能力,自组装分子动力学模拟已成为探索各种自组装体系机理的利器。
1.软物质与胶体科学:这是自组装模拟最经典的应用领域。例如,研究人员可以模拟表面活性剂或两亲性嵌段共聚物在水溶液中如何自发形成球形胶束、棒状胶束、囊泡(脂质体)等不同形貌的聚集体。
通过改变分子结构、浓度、温度或溶剂性质,模拟可以预测并解释这些形貌转变的内在原因,为设计具有特定功能的药物递送载体或纳米反应器提供理论指导。
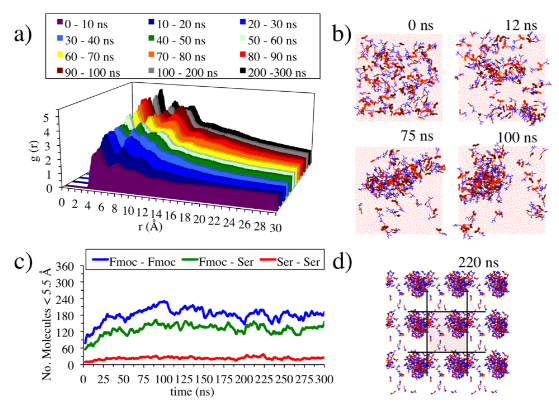
图3有机分子自组装模拟结果,DOI: 10.1039/c8cs00040a
2.生物分子体系:生命过程本身就是一部精妙的自组装史诗。MD模拟被广泛用于研究生物大分子的自组装行为。一个典型的例子是阿尔茨海默病相关的β–淀粉样蛋白(Aβ)肽段的聚集。
通过模拟,科学家可以观察单个Aβ肽段如何从无规线团逐步聚集成寡聚体,并最终形成致病的β–折叠纤维结构,从而揭示其聚集的早期关键步骤和可能的药物干预靶点。此外,病毒衣壳的组装、蛋白质复合物的形成、细胞膜上脂筏的动态行为等复杂生命过程,也都可以通过自组装模拟进行深入探索。
图4环肽的自组装模拟结果,DOI: 10.1039/c8cs00040a
关键技术与挑战
尽管自组装MD模拟功能强大,但其成功实施也面临着一系列技术挑战,其中最核心的是时间尺度、空间尺度和力场精度之间的矛盾。
1.时间与空间尺度的挑战与对策:自组装通常是一个缓慢的过程,发生在微秒(μs)到毫秒(ms)甚至更长的时间尺度上,而一个模拟体系可能包含数十万甚至上百万个原子。使用传统的全原子(All-Atom, AA)模型进行如此长时间、大体系的模拟,计算成本极高,甚至超出了当前超级计算机的能力范围。
为了克服这一瓶颈,粗粒化(Coarse-Graining, CG) 模型应运而生。CG模型通过将一组原子(如一个氨基酸残基、一个溶剂分子团)简化为一个“超级粒子”或“珠子”,显著减少了系统的自由度,使得模拟的时间步长可以大幅增加,从而能够以可接受的计算成本达到更长的时间和更大的空间尺度。
著名的CG力场如MARTINI,已在生物膜和聚合物自组装研究中取得了巨大成功。此外,元动力学(Metadynamics)等增强采样技术也可以通过引入偏置势能,加速系统跨越能垒,从而在更短的模拟时间内探索到自组装的发生。
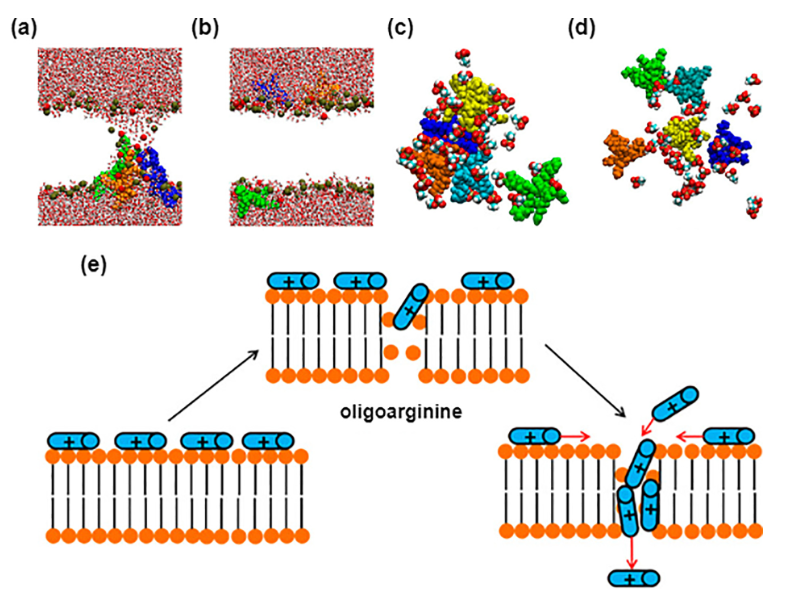
图5CPPs的自组装及其与脂质双分子层的相互作用模式,DOI:10.1063/5.0142302
2. 力场的准确性与参数化:模拟结果“准不准”,关键看力场。对于标准的生物分子和常见有机溶剂,已有许多经过充分验证的力场(如CHARMM, AMBER, OPLS-AA)可供使用。然而,当研究涉及非标准小分子(如药物、染料、新型有机配体)时,往往缺乏现成的精确参数。
这时就需要进行力场参数化,即通过高精度的量子化学计算(QM)来获得分子的键合参数和原子电荷等信息,并将其拟合到力场的函数形式中。特别是对于一些新兴的自组装体系,如含氟化合物或金属配合物,开发专门的力场参数是保证模拟可靠性的前提。
小结
自组装分子动力学模拟作为一座连接微观分子世界与宏观材料功能的桥梁,其重要性日益凸显。它不仅为我们提供了在原子级别上“设计”和“导演”分子自组装过程的能力,也正在不断推动新材料的发现和对生命奥秘的理解。
尽管在时间尺度、力场精度和多尺度耦合方面仍面临挑战,但随着计算能力的持续增长以及粗粒化、增强采样和机器学习等新方法的不断涌现,我们相信,自组装分子动力学模拟将在未来的科学探索中扮演越来越关键的作用。