

反应力场(Reactive Force Field, ReaxFF)是一种基于原子间键级(Bond Order)动态变化的分子力场,通过实时计算键的形成与断裂过程,能够模拟化学反应中的原子重排和电荷转移,突破了传统力场只能描述固定原子连接方式的局限。
它结合量子力学(QM)的准确性与分子力学(MM)的计算效率,广泛应用于燃烧、催化、材料降解等动态反应过程的模拟。与经典力场相比,ReaxFF通过引入键级函数和电荷平衡模型,可自动识别化学键的断裂与生成,在保持原子数不变的前提下模拟复杂化学反应网络。
ReaxFF力场的原理与机制
ReaxFF(Reactive Force Field)是由Adri van Duin与William Goddard团队于2001年首次提出的反应力场,旨在解决大规模反应性化学体系(如数千原子)的分子动力学模拟难题。
其核心创新在于引入 键序(Bond Order, BO) 作为动态变量,通过连续函数描述化学键的形成与断裂过程。具体而言,键序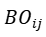
 由原子间距 决定:
由原子间距 决定:
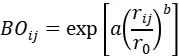
其中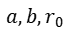 为元素相关参数。键序进一步与键能直接关联:
为元素相关参数。键序进一步与键能直接关联:
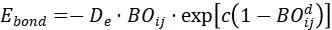
这一设计确保键能在断裂时平滑趋近于零,避免了传统力场在断键时的能量突变。例如,当两个碳原子距离增大至断键阈值时,从1.0(单键)连续降至0,键能随之衰减至零,同时依赖键序的键角能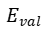 和扭转能 也同步消失,实现键断裂的物理合理。
和扭转能 也同步消失,实现键断裂的物理合理。
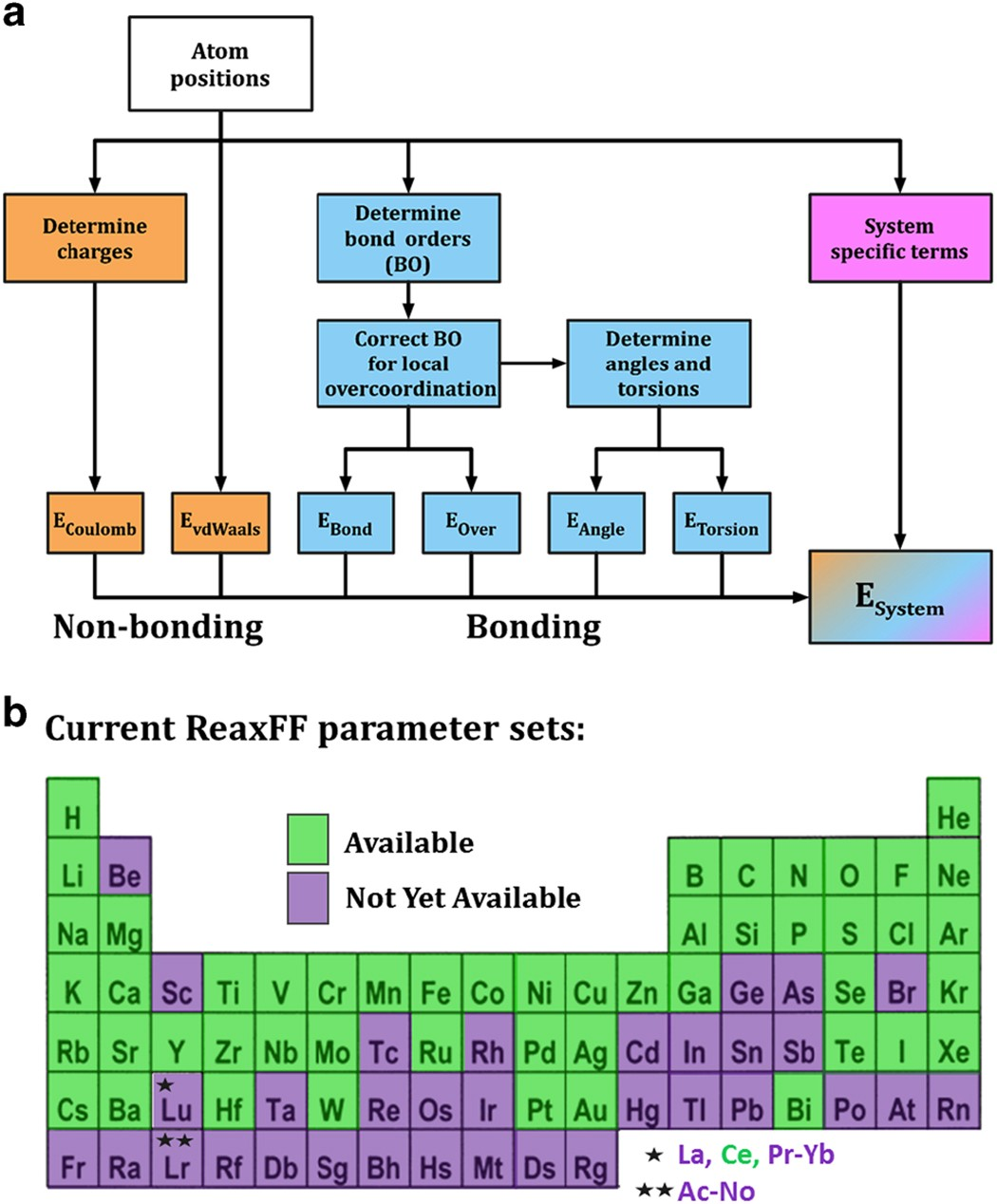
DOI:10.1038/npjcompumats.2015.11
非键相互作用的独特处理是另一关键创新。ReaxFF对所有原子对(无排除规则)计算屏蔽的库仑势与范德华势:
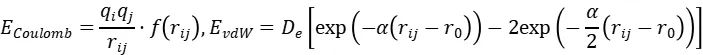
屏蔽函数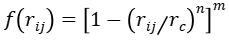 在短程()时强制势能恒定,避免数值发散。这种设计使ReaxFF能同时描述共价键断裂与长程静电作用(如离子晶体),突破了传统力场(如AMBER/CHARMM)的固定拓扑限制。
在短程()时强制势能恒定,避免数值发散。这种设计使ReaxFF能同时描述共价键断裂与长程静电作用(如离子晶体),突破了传统力场(如AMBER/CHARMM)的固定拓扑限制。
参数化方法与化学键机制
ReaxFF参数化依赖量子化学数据(如键解离能、过渡态几何构型)的精细拟合。以镁盐水合物为例,其参数通过以下步骤获得:训练集构建:结合[Mg(H₂O)ₙ]²⁺团簇的量子化学能量、晶体结构数据及振动光谱。
优化算法:采用蒙特卡洛或遗传算法,最小化ReaxFF与量子化学能量的均方差。典型参数集约100个,涵盖键序衰减指数、电荷极化率等。可转移性验证:将镁参数迁移至MgO表面水吸附模拟,对比DFT的吸附能误差需。
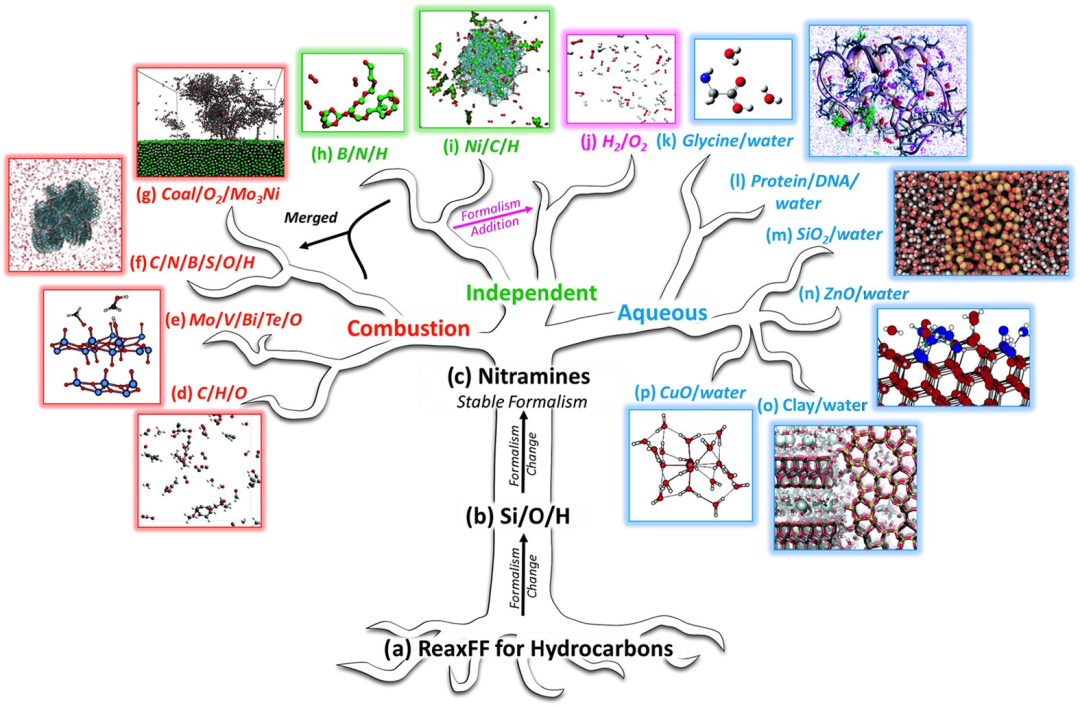
DOI:10.1038/npjcompumats.2015.11
键序计算进一步细分为σ、π、ππ三种共价键贡献:
σ键序:沿键轴方向轨道重叠,主导单键(BO=1.0)。
π键序:p轨道侧向重叠,形成双键(BO=2.0)。
ππ键序:d轨道参与,见于三键(BO=3.0)。
这种分解使ReaxFF能区分乙烯(C=C, π键)与乙炔(C≡C, ππ键)的反应活性差异,在烃类氧化模拟中成功预测丙烯活性高于苯(因C-H键能更低)。
应用场景
烃类氧化与燃烧机制
ReaxFF在高温氧化模拟中展现独特优势。以甲烷/氧气混合体系为例:
引发阶段:O₂夺取CH₄的H形成HO₂•与CH₃•,能垒26.4 kcal/mol(实验值≈27 kcal/mol)。
链式反应:CH₃•与O₂生成甲醛,进一步氧化为CO/H₂O。
产物分布:贫燃条件(氧过量)主要生成CO₂,富燃条件则积累CO与未燃烃。模拟中观察到邻二甲苯在800K时苯环开裂形成烯丙基自由基,而苯需1000K以上才反应,与C-H键强度趋势一致。
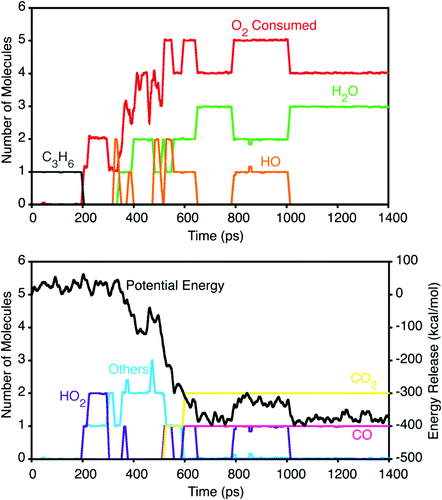
DOI:10.1021/jp709896w
异相催化界面反应
ReaxFF对 钒氧化物催化剂(V₂O₅) 的研究揭示表面氧空位的关键作用:
丙烷脱氢:V⁵⁺位点吸附C₃H₈,C-H键断裂形成丙烯,能垒1.2eV(QM计算1.3eV)。
氧再生机制:气相O₂填充氧空位,伴随电荷转移V⁴⁺→V⁵⁺。此类模拟成功预测V-O-C键角在过渡态时从120°弯曲至95°,体现ReaxFF对几何畸变的敏感性。
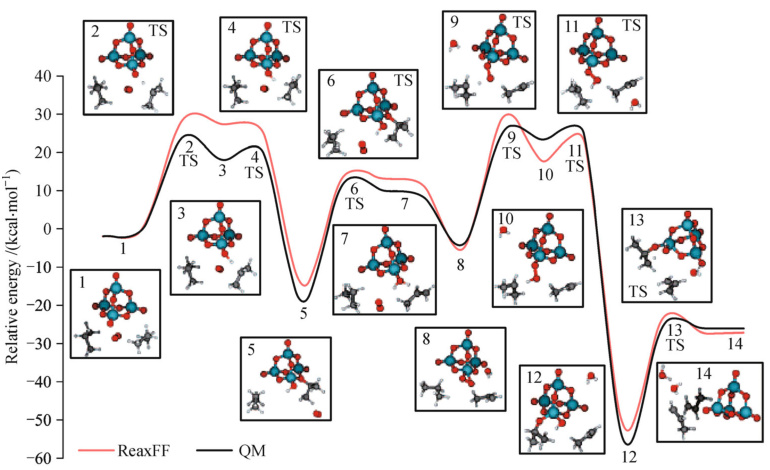
DOI 10.1007/s11705-015-1545-z
生物分子水解与材料合成
在DNA磷酸二酯键水解中,ReaxFF模拟Zn²⁺催化机制:亲核进攻:水分子OH⁻攻击磷酸磷原子,形成五配位过渡态。
键断裂:P-O₃键拉长至2.1 Å时断裂(QM值2.0 Å),能垒误差。对碳纳米管/聚合物复合材料的模拟显示,环氧树脂在CNT表面交联时,C-O键优先在五元环缺陷处形成(键序累积速率快30%)。
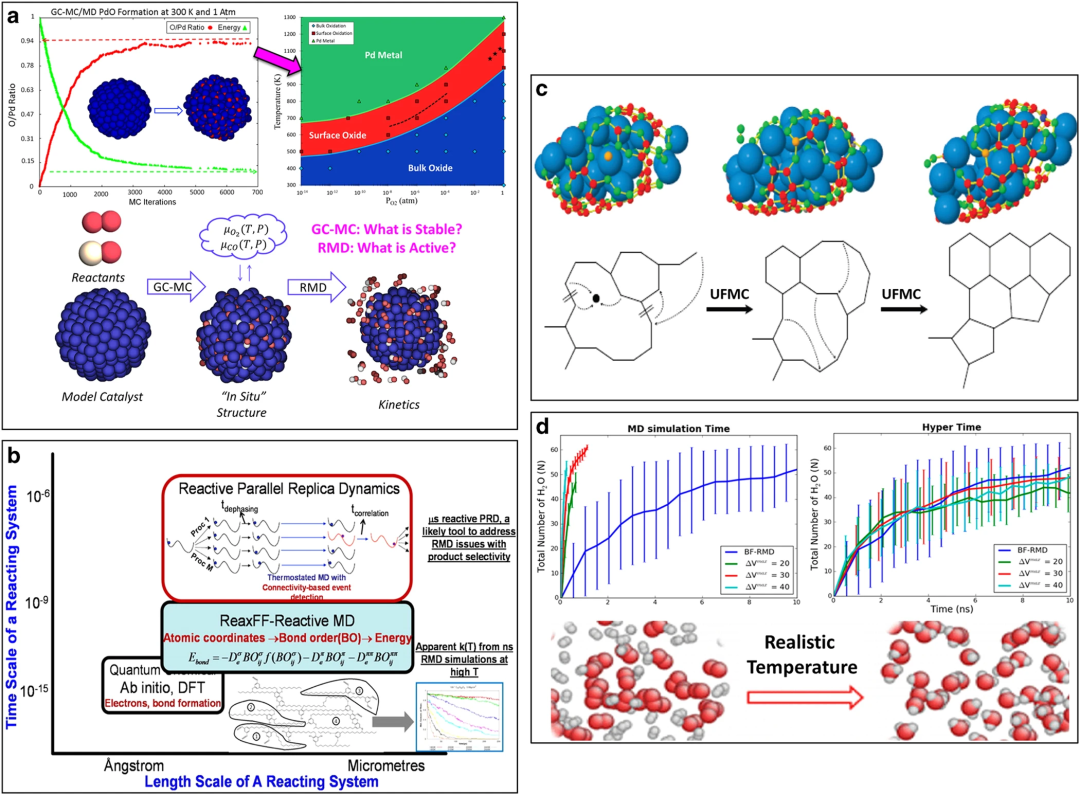
DOI:10.1038/npjcompumats.2015.11
与传统力场的对比分析
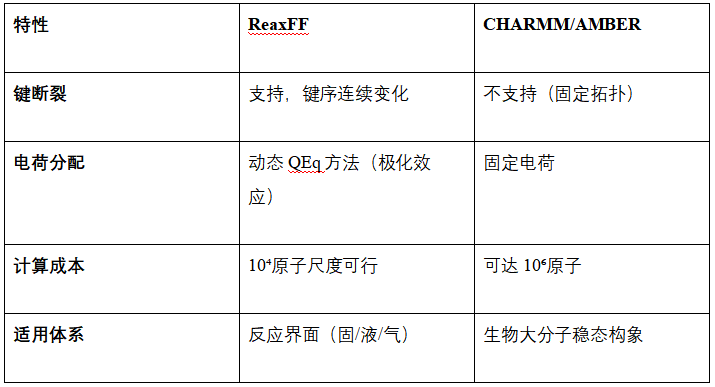
例如在DNA冲击波损伤模拟中,CHARMM无法描述磷酸二酯键断裂,而ReaxFF揭示冲击波传递时键序的位点优先断裂,与实验观测的DNA碎片化一致。而AMBER在β-肽折叠模拟中虽能采样314螺旋构象,但忽略质子转移导致的构象翻转。
总结
ReaxFF通过键序的动态耦合与非键相互作用的连续屏蔽,实现了化学反应的高效分子动力学模拟。其在燃烧科学中精准捕捉烃类氧化路径,在异相催化中揭示表面空位作用,在生物材料中解析水解机制,已成为连接量子化学与传统分子力场的桥梁。尽管参数化复杂性与计算成本仍是挑战,自适应采样等算法的引入正推动其向更大体系、更高精度演进。