高斯软件是一款在计算化学领域占据重要地位的专业软件。自 1970 年由约翰・波普尔等人发布第一个版本以来,经过多年的持续研发和改进,已经发展成为功能强大、应用广泛的计算化学软件包。
它的名称源于在分子电子结构计算中采用高斯函数来近似描述电子轨道,这种方法能够有效地提高计算效率,适应当时计算机硬件的有限性能。
高斯软件的发展历程见证了计算化学领域的不断进步,其每一次版本更新都带来了新的功能和改进,使其在处理复杂化学问题时的能力不断增强。
有机化学:高斯软件在有机化学研究中有着广泛的应用。它可以帮助化学家预测有机反应的产物,通过计算反应路径和过渡态结构,深入了解反应的机理。
例如,在研究亲核取代反应、加成反应等常见有机反应时,高斯软件能够提供详细的分子结构变化信息,解释反应的选择性和立体化学特征。
此外,对于有机分子的结构优化和稳定性分析也是高斯软件的强项,它可以确定分子在不同条件下的最稳定构象,为有机合成路线的设计提供理论依据。
无机化学:在无机化学领域,高斯软件可用于研究金属配合物的结构与性质。通过计算配合物的电子结构,包括轨道能量、电荷分布等,可以解释配合物的光谱性质、磁性以及反应活性。
例如,对于过渡金属配合物的催化性能研究,高斯软件可以帮助揭示催化剂与底物之间的相互作用机制,为设计高效的无机催化剂提供指导。
同时,对于无机材料的晶体结构优化和性质预测也是高斯软件的应用方向之一,有助于探索新型无机功能材料。
物理化学:高斯软件在物理化学研究中发挥着重要作用。它可以计算分子的热力学性质,如焓、熵、自由能等,为化学反应的热力学分析提供数据支持。
通过计算反应的活化能和反应速率常数,还能深入研究化学反应的动力学过程。
此外,在研究分子间相互作用,如氢键、范德华力等方面,高斯软件也能提供准确的计算结果,帮助理解物质的聚集态结构和物理性质。
电子结构与光学性质研究:高斯软件能够精确计算材料的电子结构,包括能带结构、态密度等重要信息。通过对这些结果的分析,可以了解材料的导电性、半导体特性以及光学吸收等性质。
例如,在研究新型半导体材料时,高斯软件可以预测材料的禁带宽度,为调整材料的光学和电学性能提供理论指导。
对于发光材料,它可以计算分子的激发态性质,解释材料的发光机制,从而助力优化材料的发光效率。
材料结构与性能关系研究:通过模拟材料的晶体结构和缺陷形成能,高斯软件可以研究材料的结构稳定性和性能之间的关系。
例如,在研究金属合金材料时,它可以分析合金元素对材料晶体结构和力学性能的影响。对于陶瓷材料,能够研究晶格缺陷对材料电学和力学性能的作用。
此外,在研究高分子材料时,高斯软件可以模拟高分子链的构象和相互作用,为材料的加工和性能优化提供依据。
生物大分子结构与功能研究:高斯软件可用于研究蛋白质、核酸等生物大分子的三维结构与功能关系。通过计算生物大分子的构象变化、折叠过程以及与配体的相互作用,可以深入了解生物分子的活性位点和作用机制。
例如,在研究酶的催化机制时,高斯软件可以模拟酶与底物的结合过程,揭示催化反应的关键步骤。
对于蛋白质 – 核酸相互作用的研究,它可以帮助确定相互作用的位点和方式,为理解基因表达和调控机制提供理论支持。
药物分子设计与优化:在药物化学领域,高斯软件是药物分子设计和优化的重要工具。它可以计算药物分子与生物靶点的相互作用,如结合模式、结合能等,从而指导药物分子的设计和筛选。
通过对药物分子的结构进行优化,提高其与靶点的亲和力和特异性,同时降低对非靶标蛋白的作用,从而提高药物的活性和安全性。
此外,高斯软件还可以预测药物分子的药代动力学性质,如吸收、分布、代谢和排泄等,为药物的临床研究提供参考。
1. 下载软件:首先,从高斯软件官方网站或授权的软件经销商处获取适用于 Windows 系统的高斯软件安装包。通常,安装包是一个压缩文件,大小根据软件版本和包含的组件不同而有所差异。
2. 解压安装包:将下载的压缩文件解压到本地磁盘的指定目录。建议选择一个空间充足且易于访问的位置,如 “C:Program FilesGaussian” 或其他自定义的文件夹。
3. 运行安装程序:进入解压后的文件夹,找到名为 “setup.exe” 或类似名称的安装程序文件,双击运行它。安装程序将启动高斯软件的安装向导。
4. 选择安装选项:在安装向导的第一步,通常会要求用户选择安装语言。选择您熟悉的语言后,点击 “下一步”。接下来,您将看到许可协议页面,仔细阅读并接受许可协议后,点击 “下一步”。
然后,您可以选择安装类型,如 “典型安装”、“自定义安装” 等。典型安装会安装高斯软件的基本组件和常用工具,适合大多数用户;自定义安装则允许用户根据自己的需求选择安装特定的组件和功能。
5. 设置安装路径:在选择安装类型后,安装向导会要求用户指定软件的安装路径。您可以使用默认的安装路径,也可以点击 “浏览” 按钮选择其他位置。确保安装路径所在的磁盘有足够的空间来容纳高斯软件及其相关文件。
6. 配置环境变量:安装过程中,安装程序可能会自动配置系统环境变量,以便您能够在任何位置方便地运行高斯软件。
如果安装程序没有自动配置,您需要手动将高斯软件的安装目录添加到系统的 “PATH” 环境变量中。这样,您就可以在命令提示符或其他终端中直接输入高斯软件的命令来启动程序。
7. 完成安装:完成上述设置后,点击 “下一步”,安装程序将开始复制文件并安装高斯软件。安装过程可能需要一些时间,具体取决于您的计算机性能和安装组件的数量。
安装完成后,您可以选择是否创建桌面快捷方式或开始菜单快捷方式,以便快速访问高斯软件。最后,点击 “完成” 按钮退出安装向导。
1. 获取安装文件:从高斯软件官方网站下载适用于 Linux 系统的安装文件。通常,安装文件是一个.tar.gz 格式的压缩包,包含了高斯软件的源代码和安装脚本。
2. 解压安装文件:使用命令行工具,如 “tar -zxvf [安装文件名].tar.gz”,将压缩包解压到指定的目录。建议将其解压到一个专门的软件安装目录,如 “/opt/gaussian” 或用户自定义的目录。
3. 配置安装环境:进入解压后的目录,查看安装文档或 README 文件,了解软件的安装要求和配置步骤。
通常,需要设置一些环境变量,如 “GAUSS_EXEDIR”,指定高斯软件可执行文件的目录;“GAUSS_SCRDIR”,指定临时文件和计算结果的存储目录等。
您可以将这些环境变量设置添加到您的 shell 配置文件中,如 “.bashrc” 或 “.zshrc”,以便在每次登录时自动加载。
4. 编译安装:根据安装文档的指导,运行安装脚本或执行编译命令来安装高斯软件。这可能涉及到一些编译选项和依赖库的安装。
如果系统缺少某些依赖库,您需要先使用包管理工具(如 apt、yum 等)安装相应的库。编译过程可能需要较长时间,尤其是在处理大规模计算任务或安装多个组件时。
5. 测试安装:安装完成后,运行一些简单的测试案例来验证高斯软件是否安装成功。可以使用高斯软件自带的测试文件或自己编写一个简单的输入文件进行计算。
如果计算能够正常运行并生成正确的结果,说明高斯软件已经成功安装在 Linux 系统上。
薛定谔方程:高斯软件的核心理论基础是量子力学,其通过求解薛定谔方程来描述分子体系的电子结构和能量。
薛定谔方程是量子力学的基本方程,它将分子的哈密顿算符与波函数联系起来,通过求解波函数得到分子的能量和电子分布。
对于一个包含N个电子和M个原子核的分子体系,其薛定谔方程可以表示为:Hψ = Eψ,其中H是哈密顿算符,ψ是波函数,E是分子的能量。
波函数与电子结构:波函数是描述分子中电子运动状态的数学函数,它包含了分子的电子结构信息。通过对波函数的分析,可以得到分子的电子云分布、轨道能量、电荷密度等重要物理量。
在高斯软件中,通常采用原子轨道线性组合(LCAO)的方法来近似表示分子的波函数,即将分子轨道表示为原子轨道的线性组合,从而将复杂的多电子问题转化为相对简单的原子轨道组合问题进行求解。
分子轨道的形成:分子轨道理论认为,当原子相互靠近形成分子时,原子轨道会相互重叠并重新组合形成分子轨道。分子轨道分为成键轨道和反键轨道,成键轨道上的电子有助于原子间的结合,而反键轨道上的电子则不利于原子间的结合。
高斯软件通过计算分子轨道的能量和形状,来分析分子的化学键性质和化学反应活性。例如,通过比较成键轨道和反键轨道的电子占据情况,可以判断分子的键级和稳定性。
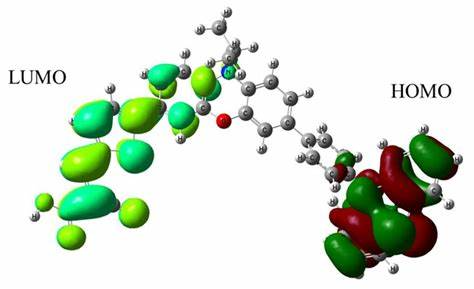
电子排布与分子性质:根据泡利不相容原理和洪特规则,电子在分子轨道上进行排布。分子的化学性质和物理性质与其电子排布密切相关。
高斯软件可以计算分子的最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)的能量,HOMO – LUMO 能隙是衡量分子化学反应活性的重要指标。能隙越小,分子越容易发生化学反应,因为电子更容易从 HOMO 跃迁到 LUMO。
此外,分子轨道的形状和对称性也对化学反应的选择性和机理有着重要影响。
电子密度泛函:密度泛函理论是高斯软件中广泛应用的一种计算方法。它将分子的能量表示为电子密度的泛函,而不是像传统的量子力学方法那样直接求解波函数。
通过对电子密度的变分原理,可以得到分子的能量和电子结构。与传统的从头算方法相比,DFT 在计算精度和计算效率上取得了较好的平衡,能够处理较大的分子体系。
例如,在研究生物大分子或材料体系时,DFT 方法可以在相对较短的时间内提供较为准确的计算结果。
交换 – 关联泛函:在密度泛函理论中,交换 – 关联泛函是描述电子之间交换和关联相互作用的关键部分。不同的交换 – 关联泛函具有不同的形式和精度,常见的有局域密度近似(LDA)、广义梯度近似(GGA)以及杂化泛函(如 B3LYP)等。
LDA 适用于均匀电子气体系,GGA 考虑了电子密度的梯度信息,而杂化泛函则将精确的交换项与密度泛函理论的关联项相结合,能够更好地描述分子的电子结构和性质。
高斯软件提供了多种交换 – 关联泛函供用户选择,用户可以根据具体的研究体系和计算要求选择合适的泛函。
原理:通过调整分子中原子的坐标,使分子体系的能量达到最小值,得到稳定的分子几何结构。这是基于量子力学中的能量最小化原理,即分子在稳定状态下具有最低的能量。
应用:可以确定分子的平衡构型,包括键长、键角、二面角等几何参数。
对于有机化合物、生物分子、材料分子等的结构研究至关重要,有助于理解分子的空间结构和性质之间的关系。
例如,在药物设计中,明确药物分子与靶点的结合模式,需要先优化药物分子的结构。
原理:基于量子化学理论,计算分子体系的总能量,包括电子能量、核 – 核排斥能等。常用的方法有密度泛函理论(DFT)、 Hartree – Fock(HF)方法等。
应用:用于比较不同分子结构或不同化学反应状态的能量高低。
在化学反应机理研究中,通过计算反应物、中间体和产物的能量,确定反应的热力学可行性和反应热。
还可以用于研究分子的稳定性,能量越低,分子越稳定。
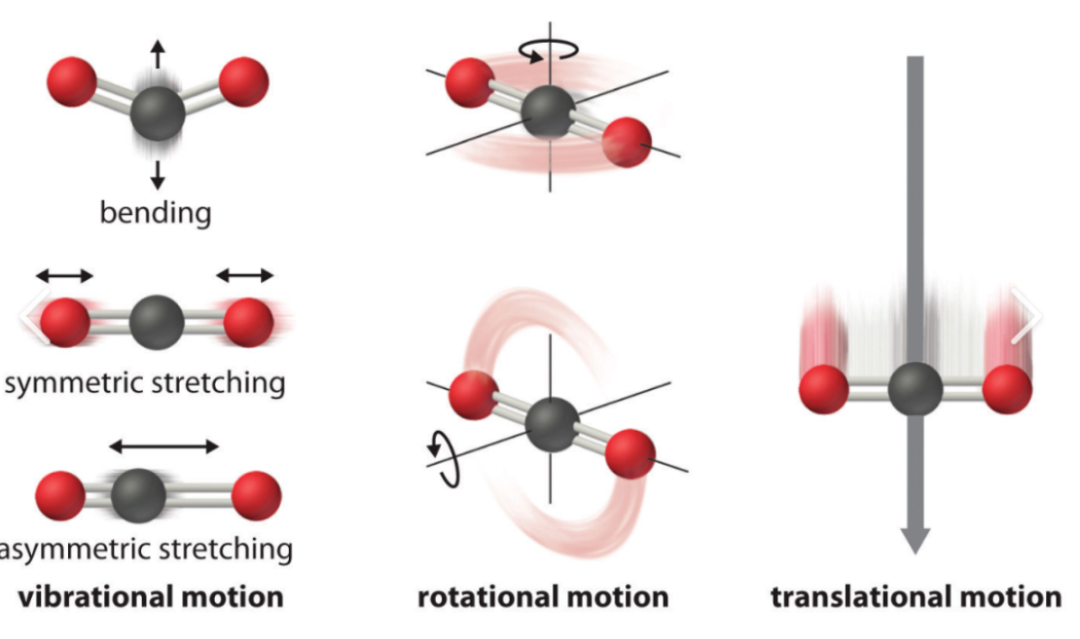
原理:通过计算分子的 Hessian 矩阵,得到分子的振动频率和振动模式。Hessian 矩阵的元素与分子能量对原子坐标的二阶导数相关,反映了分子在平衡位置附近的势能面曲率。
应用:用于确定分子的红外和拉曼光谱,为实验光谱的解析提供理论依据。
通过分析振动频率,可以判断分子的结构是否处于稳定的极小值点(所有振动频率均为正值),还是处于过渡态(有且仅有一个虚频)。
此外,还可以计算分子的热力学性质,如热容、熵等,这些性质与分子的振动模式密切相关。
原理:计算分子的电子密度分布、分子轨道能量和形状等。分子轨道理论认为,分子中的电子是在整个分子范围内运动,通过求解薛定谔方程得到分子轨道及其能量。
应用:分析分子的化学键性质,如成键轨道和反键轨道的分布,判断化学键的类型和强度。
研究分子的电子跃迁性质,如吸收和发射光谱,对于理解分子的光学性质和光化学反应机理具有重要意义。
此外,电子结构分析还可以用于研究分子的电荷分布和极化性质,为分子间相互作用的研究提供基础。
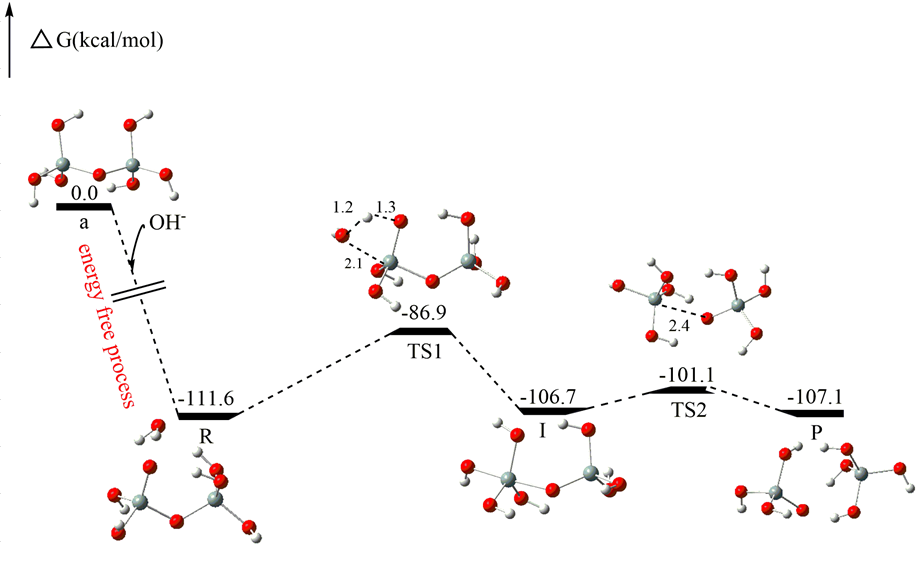
原理:通过计算反应路径上的关键中间体和过渡态的结构和能量,构建反应的势能面。利用过渡态理论和量子化学计算方法,确定反应的速率常数和反应机理。
应用:在有机合成反应中,预测反应的产物和选择性,为实验合成提供理论指导。例如,研究不对称催化反应的机理,有助于设计更高效的手性催化剂。
在生物化学领域,研究酶催化反应的机理,了解酶与底物之间的相互作用和反应过程,为药物设计和酶工程提供理论依据。
在材料科学中,研究材料表面的化学反应机理,对于材料的表面改性和催化性能的优化具有重要意义。
原理:计算分子之间的范德华力、静电相互作用、氢键等相互作用能。常用的方法有分子力学(MM)方法、量子力学 / 分子力学(QM/MM)组合方法等。
应用:在药物设计中,研究药物分子与生物靶点之间的相互作用,优化药物分子的结构,提高药物的活性和选择性。
在材料科学中,研究分子在材料表面的吸附行为和相互作用,对于理解材料的表面性质和吸附分离过程具有重要意义。
在溶液化学中,研究溶质分子与溶剂分子之间的相互作用,解释溶液的性质和化学反应在溶液中的行为。
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!