

一、原理介绍
虚拟筛选(Virtual Screening, VS)是一种借助计算机技术,从海量化合物库中寻找可能与药物靶点(通常是蛋白质受体或酶)结合的小分子的过程。
分类
1、基于受体的虚拟筛选(Structure-Based Virtual Screening, SBVS):以靶蛋白的三维结构为基础,研究靶蛋白结合位点的特征性质及与小分子化合物间的相互作用模式,依据与结合能相关的打分函数,评估蛋白和小分子化合物的结合能力,从而从众多化合物中筛选出结合模式合理、预测得分较高的化合物。
2、基于配体的虚拟筛选(Ligand-Based Virtual Screening, LBVS):利用已知活性的小分子化合物,根据其形状相似性或药效团模型,在化合物数据库中搜索匹配的化学分子结构。
筛选流程
基于受体的虚拟筛选流程
1. 靶标选择与结构获取:挑选与疾病相关的靶蛋白,通过生物信息学方法获取其三维结构和结合口袋位置,进行蛋白准备并设置对接参数,这与分子对接的处理方式一致。
2. 小分子库的准备:准备好包含数千到数百万小分子的化合物库,这些化合物可为天然产物、小分子药物或化学合成的化合物等。将文件转换为软件可识别的格式,用于分子对接筛选。
3. 对接模拟:运用分子对接方法,模拟小分子与靶标蛋白的结合模式,预测它们之间的相互作用强度。常见的对接软件有AutoDock Vina、Dock、Glide(Schrödinger)等,Schrödinger等软件有进行虚拟筛选的模块Virtual Screen Workflow。
4. 筛选与优化:对接模拟后,筛选出最有可能与靶标结合的小分子,并结合小分子的结合能、应变能、ADMET(吸收、分布、代谢、排泄、毒性)类药性质等方面的评估,进一步筛选活性分子。
5. 分子动力学模拟(可选):进一步评估活性小分子与靶蛋白动态结合的稳定性和其在体内的行为,以确定其药理学性质和生物活性。
6、小分子化合物活性验证。


图1 基于受体多精度对接方法组合的虚拟筛选示例
基于配体的虚拟筛选流程
1. 药效团模型构建:分析一个或多个活性小分子的药效特征,概括出使分子具有活性的重要药效基团特征。
2. 模型验证:构建包含已报道的活性化合物和无活性的诱饵分子的模型,验证药效团模型的有效性。
3. 筛选策略:利用构建好的药效团模型在化合物数据库中搜索含有相同特征的小分子。
4. 相似性搜索:将已知的活性化合物与数据库中化合物进行相似性计算打分,根据设定的相似性阈值,挑选出满足相似性要求的分子。
5、小分子化合物活性验证。
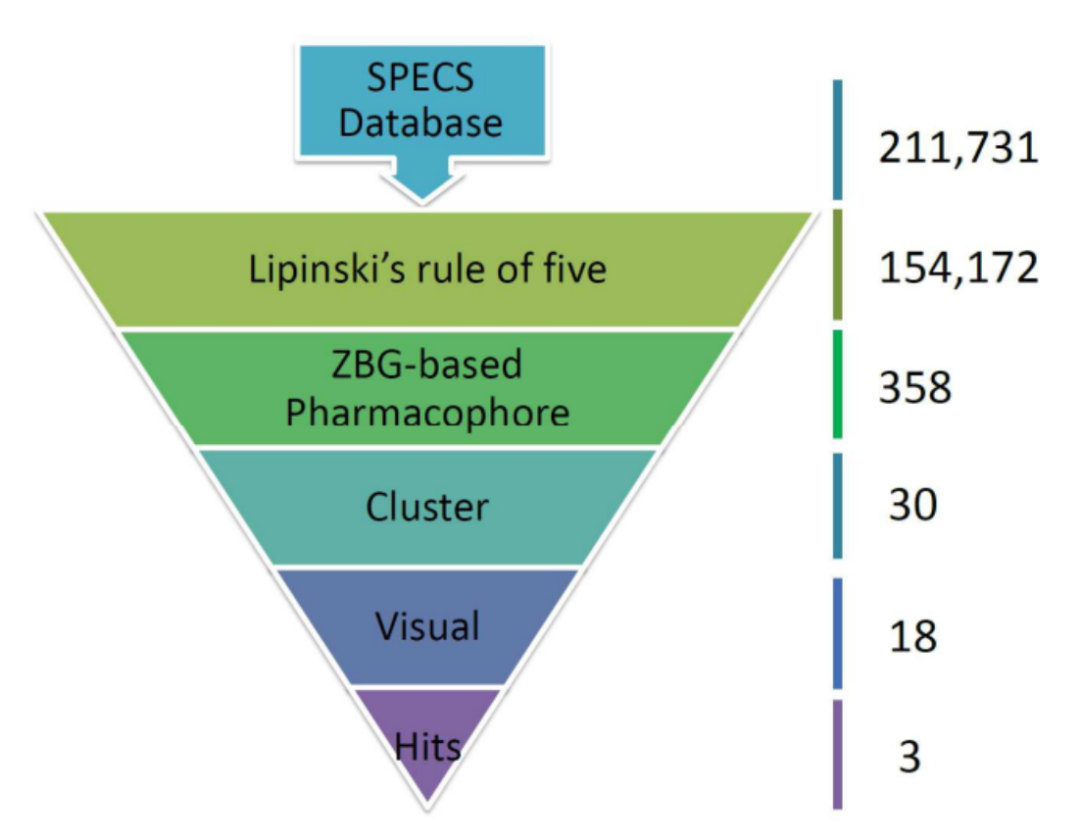
图2 基于配体的虚拟筛选示例
虚拟筛选的优势
1. 高效性:可在短时间内筛选大量化合物,大幅减少实验筛选的化合物数量,缩短新药发现周期,大大减低高通量筛选的时间、人力、经济成本。
2. 降低成本:降低药物研发成本,提高先导化合物的发现效率。
3. 组合方法灵活多变:可根据研究内容设置多种组合筛选方法。
二、虚拟筛选的应用
1. 靶标识别与验证:通过虚拟筛选,可从现有蛋白质数据库中筛选出与某种疾病相关的靶标,并通过实验进一步验证其可行性。
2. 小分子化合物的筛选与优化:通过筛选化合物库,快速找到与靶标蛋白相互作用的小分子,这些分子可能是新的候选化合物,或者是已知药物的潜在新用途。
3. 药物研发:虚拟筛选已成为目前最有潜力的药物开发工具,成功辅助药物设计的案例逐年增多。
三、顶刊解读

近期开发并应用的通过结构-活性关系(XSAR)集来寻找适合卤键形成的特定氨基酸(XB热点)的分子建模方法,已被应用于5-羟色胺7受体(5-HT7R)的研究。在此研究中,S5x42(特定氨基酸)作为虚拟筛选的一个约束条件。研究者通过虚拟筛选,从超过800万种的市售化合物中最终筛选出63个XSAR集(共156种化合物),并使用体外实验对其与5-HT7R的亲和力进行检测。结果预测的卤代衍生物对5-HT7R的亲和力高于其未取代类似物的准确性为68%。
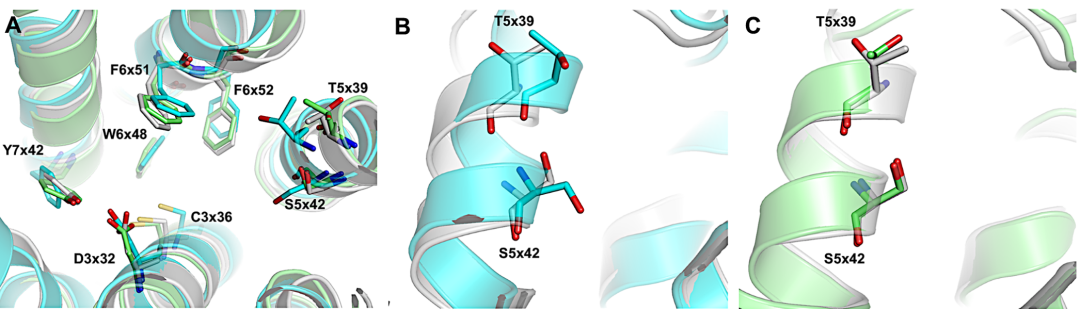
图1 (A)晶体的5-HT7受体结合位点(灰色)与先前研究中使用的两种同源模型(青色和绿色)的比较。
将同源模型结构与5-HT7受体晶体结构进行叠加(图1A)显示,与配体结合的关键氨基酸的位置非常吻合。尽管存在差异,S5x42和T5x39氨基酸仍被XSAR库对接识别为5HT7实验结构中卤素键的优选位点,用于虚拟筛选研究,而且结果表明这些细微差异并不会对筛选结果造成影响,也证实了同源建模方法的准确性。
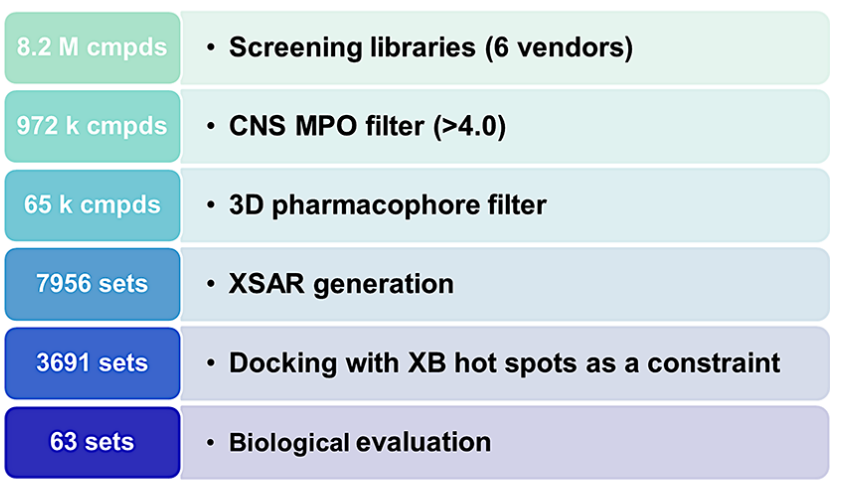
图2 研究采用的虚拟筛选方案。
为了合理化该研究提出的实验设计,一个包含超过800万种化合物的库在虚拟筛选过程中数目逐步被筛选(如图2所示)。由于5-HT7受体属于中枢神经系统药物的药理学靶点,因此第一步的筛选是基于一组六个基本分子描述符构建的中枢神经系统药物性模型。经过筛选,大约有972,000种化合物的CNS成药性指数至少为4.0,从而进入下一阶段。在虚拟筛选的下一阶段,使用了两种不同的三维药效团模型(它们基于结构不同的5-HT7R拮抗剂构建)。筛选标准是进入下一阶段的化合物必须完全满足至少其中一个模型的所有规则,即化合物能够映射到模型的所有药效团特征。这使得化合物数目进一步减少到约65,000个结构。
表1 从3691个XSAR集的对接分析中得到的所有卤键中氨基酸的归一化贡献(按受体类型)
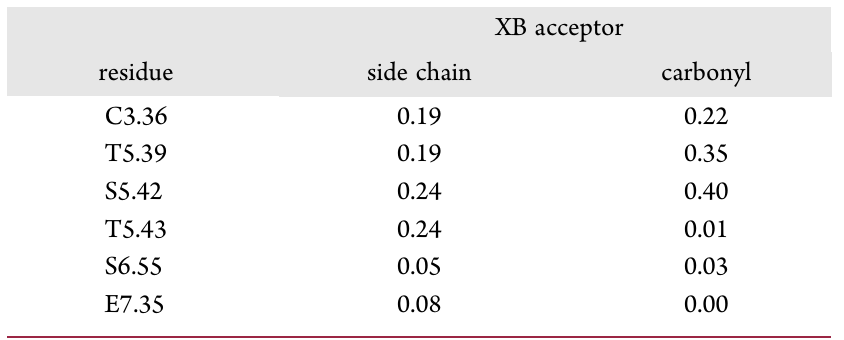
在筛选后的化合物库中,研究者通过对接和分析进一步筛选潜在的活性小分子。首先,获得7956个XSAR数据集(包含母体结构及其卤代衍生物),并将其对接到通过IFD(诱导契合对接)程序识别的所有5-HT7R构象中。利用结构相互作用指纹图(SIFt)分析结合模式,剔除不符合5-HT7R-配体关键相互作用的数据集。在满足结合模式标准的XSAR集中,进行卤素键形成分析,最终有3691个数据集满足几何标准(XB长度
为了确定最终购买的化合物,仅选择与S5x42形成卤素键的XSAR集(3113个集),并剔除未取代化合物与卤代衍生物结合能为负的集。最终选择63个集(156种化合物)。预测的卤代衍生物对5-HT7R的亲和力高于其未取代类似物的准确性为68%,研究表明卤键可作为虚拟筛选的约束条件。
https://doi.org/10.1021/acs.jmedchem.4c00816