

一、原理介绍
分子对接是模拟小分子配体与生物大分子受体结合过程的方法。它基于受体结构信息,通过计算预测配体在受体结合位点的最佳位置和方向(最佳构象),使配体-受体相互作用能量最低、结构最稳定。分子对接的核心是空间识别(结合模式)和能量识别(结合强度),空间匹配是相互作用的前提,能量匹配是稳定结合基础。


图1 分子对接的理论基础。
基本流程(以Schrödinger软件为例)
1、蛋白获取与准备
分子对接的基础是获取蛋白的三维结构模型,常用方法包括X射线晶体学、冷冻电镜技术、核磁共振波谱法等。对于无晶体模型的蛋白质,可通过同源建模或AlphaFold等方法预测蛋白的晶体结构。一般直接从PDB数据库(https://www.rcsb.org/)下载已经解析的蛋白晶体,蛋白准备是分子对接前的关键步骤,包括删除无关溶剂小分子、助晶分子等,检查保守水分子(位于结合口袋附近)、添加氢原子和电荷、补充缺失部分、检查氨基酸残基的质子化状态以及能量优化等。这些步骤通常在PyMOL、Schrödinger、Discovery Studio等软件中完成,确保蛋白质结构合理化,为分子对接提供准确的输入文件。

图2 PDB蛋白晶体数据据库。
2、配体获取与准备
配体分子的获取与准备也关键步骤之一。可以通过从免费的小分子库下载,如:
PubChem(https://pubchem.ncbi.nlm.nih.gov/)
ZINC(https://zinc15.docking.org/tranches/home/)
ChEMBL(https://www.ebi.ac.uk/chembl/)
ChemSpider(https://www.chemspider.com/search)
DrugBank(https://go.drugbank.com/)等
或者通过ChemDraw等绘图软件进行构建。配体准备前,配体需构型优化以获取低能量稳定构象,常用Gaussian软件完成。配体准备包括格式转换、添加氢原子和电荷、调整质子化状态,以及构象搜索。Schrödinger的LigPrep模块可在配体准备时生成构象,而AutoDock在对接时生成构象。
图3 PubChem生物活性小分子数据库。
3、对接参数设置
对接参数的设置是确保获得准确可靠结果的关键步骤。对接参数设置通常包括定义受体(包括对特定氨基酸残基的旋转、限制等)、定义结合位点、配体分子设置(包括对接中的核心活动区域和最大活动区域、不可活动区域等)、对接参数设置(对接精度、打分函数等)、定义结果输出的格式等。
4、对接打分
对接打分,即在上述准备完成后,将配体分子与受体的结合口袋进行对接,利用选择的打分函数,对上述的对接结果进行打分,从而对受体-配体结合情况进行评估。常用的打分函数包括四类,即基于力场(AutoDock、AutoDock Vina等)、基于知识(PMF等)、基于经验(Schrödinger等)以及基于机器学习(RF-Score等)的打分函数。尽管打分函数可以快速评估配体与受体的结合情况,但其结果存在一定的局限性,需要更准确的结合能评估。
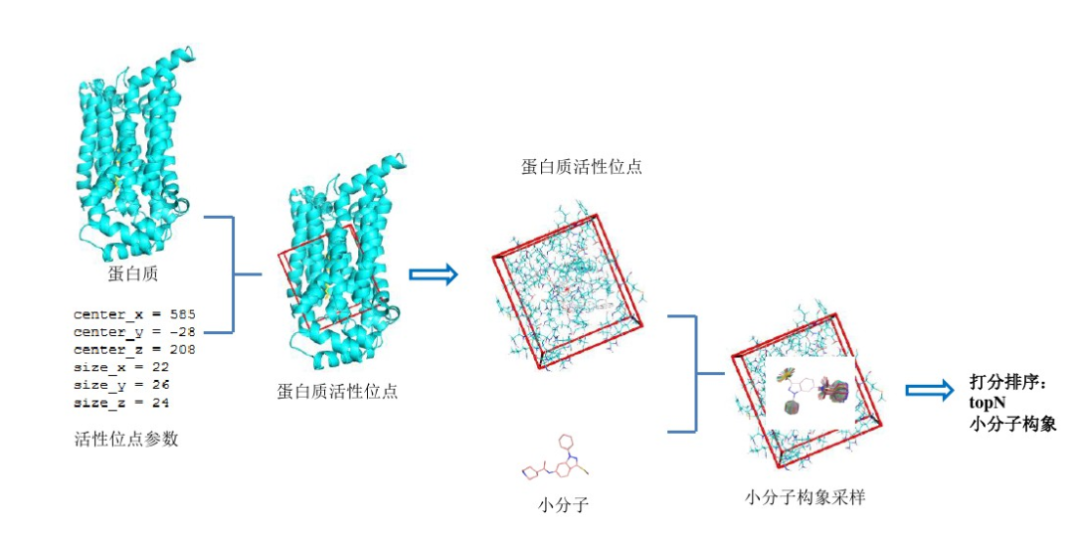
图4 AutoDock进行分子对接的示意图。
5、结合能计算
相对于打分函数,配体-受体结合能计算通过更精确的能量评估,能够更准确地预测配体-受体的结合情况,应用中一般会结合对接打分和结合能计算结果,进行综合评估。
6、应变能计算
应变能计算是分子对接中评估配体从溶液最优构象转变为与蛋白结合构象时的能量代价。由于配体在结合过程中可能产生应变能,即使结合能为负值,总能量也可能高于溶液中的能量,表明结合过程需要付出过高的能量,可能不具有实际的生物化学意义。因此,应变能计算是剔除能量过高、不合理构象的关键步骤,有助于筛选出更合理、更稳定的结合构象。
7、作用模式分析(可视化分析)
作用模式分析是通过三维视图清晰展示受体与配体之间的相互作用,涵盖氢键、范德华力、静电作用、疏水作用、π-π堆叠、π-阳离子、共价作用和卤键等多种作用模式。在研究中,通常以已知药物分子为参照,若设计或筛选的化合物不仅保留了关键残基的作用模式,还引入了新的作用模式,并且展现出比参照药物更好的亲和力,则说明这些化合物可能具有更高的潜在活性。
二、分子对接的应用
1. 小分子-靶点结合评估
结合模式预测:通过分子对接预测配体与受体的最佳结合构象,并进行打分评估。
结合能预测:评估配体与受体的结合稳定性,预测结合能。
2. 药物设计与发现
共价/非共价化合物的优化:通过分子对接优化化合物与靶点的结合亲和力。
虚拟筛选:大规模筛选化合物库,快速识别与靶点结合良好的候选化合物。
靶标垂钓:利用反向对接技术,寻找化合物的潜在靶点。
3. 酶机理研究
探究酶底物特性:分析酶与底物的结合模式,揭示酶的催化机制。
工程酶改造:设计和优化工程酶,提高其催化效率和选择性。
4. 分子相互作用研究
药物相互作用(DDI):分析药物之间的竞争结合,预测潜在的药物相互作用。
蛋白相互作用(PPI):研究蛋白之间的相互作用,调控蛋白功能。
蛋白-核酸相互作用:研究生物分子的功能和机制。
三、常用软件
开源软件
AutoDock:经典的分子对接工具,预测结合模式。
AutoDock Vina:改进版,速度快、精度高,支持多线程,适用于高通量虚拟筛选。
Vina-GPU 2.0:基于GPU加速,显著提高计算速度,适合大规模分子对接。Vinardo:优化打分函数,提高对接精度。
QuickVina 2:精准加速,提高对接效率。
QuickVina-W:快速盲对接,适用于未知结合位点的筛选。
Smina:支持自定义打分,提高打分和能量最小化性能。
SwissDock:基于Web平台,用户友好,支持多种输入方式。
LeDock:对接速度快、准确度高,适合快速筛选。
ZDOCK:专注于蛋白质-蛋白质相互作用的对接。
商业软件
Glide (Schrödinger):提供标准精度(SP)和高精度(XP)模式,对接精度高,视图功能强大,支持多精度组合虚拟筛选。
LibDock (Discovery Studio):提供多种打分函数,适用于药物设计和虚拟筛选。
GOLD:提供多种打分函数,广泛应用于药物发现。
基于深度学习的分子对接模型
Gnina:基于卷积神经网络(CNN)对配体进行评分和优化,适合高精度对接。
DeepDocking:利用深度学习模型预测对接分数,适合大规模分子对接。
四、顶刊解读


图5 新型香豆素类AR拮抗剂的设计。(A) 1(浅蓝色)与AR的结合口袋(青色)(PDB ID: 5JJM)的分子对接分析。(B)化合物1的合理修饰。
雄激素受体(AR)是前列腺癌的关键驱动因子,但AR拮抗剂的获得性耐药限制了其疗效。研究者发现香豆素衍生物1能破坏AR配体结合域二聚体,但其口服生物利用度较差。通过分子对接模拟,研究者对化合物1与AR 结合口袋(PDB ID: 5JJM)的结合模式进行分析,发现香豆素的羰基与Gln711形成关键氢键,4-Br-苯基嵌入疏水空腔。基于分子对接研究,研究者对香豆素部分和醚连接体进行系统修饰,得到两个系列的目标化合物1a−1m和2a−2e。构效关系(SAR)研究显示,4-CF₃取代香豆素和氨基连接体是抑制AR的最佳基团。进一步优化末端苯环和氨基连接子,得到化合物3a−3l和4a−4h,其中化合物4a为有进一步开发潜力的AR抑制剂候选化合物。
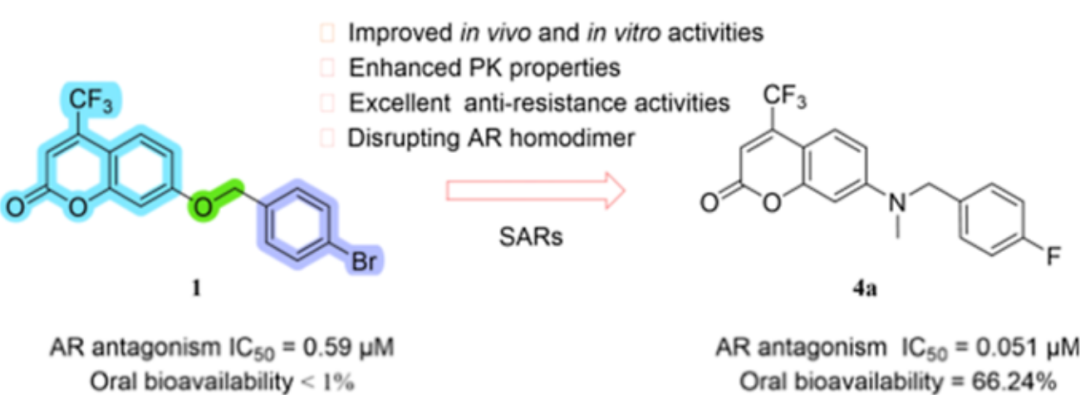
图6 基于分子对接指导香豆素衍生物1修饰得到的化合物4a。
https://doi.org/10.1021/acs.jmedchem.4c01752