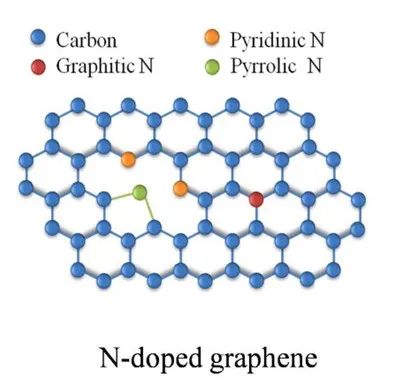
N掺杂石墨烯是通过将氮原子引入石墨烯晶格中,替代部分碳原子或形成特定功能化结构,从而调控其电子、光学和化学性质的功能化材料。
理论计算(如密度泛函理论,DFT)表明,氮的掺杂会显著改变石墨烯的能带结构、载流子浓度和磁性等性质。
例如,石墨氮(graphitic N)通过替代碳原子形成sp²杂化结构,引入额外电子导致n型导电,而吡啶氮(pyridinic N)和吡咯氮(pyrrolic N)则因局域化缺陷态呈现p型或n型行为。
通过分析氮原子的键合方式,将N掺杂分为以下主要类型:
石墨氮(Graphitic N):直接替代碳原子进入六元环,贡献1个额外电子到共轭体系,保持平面性但引入n型掺杂效应,且能保持较高的导电性。
DOI: 10.1103/PhysRevLett.116.126805
吡啶氮(Pyridinic N):位于石墨烯边缘或缺陷处,形成两个相邻的sp²键。理论计算表明其具有更高的形成能,但高浓度时更稳定。紫外光电子能谱(UPS)显示其降低功函数,增强π态密度。
吡咯氮(Pyrrolic N):位于石墨烯边缘或缺陷处,贡献2个p电子与相邻碳原子形成两个σ键,剩余1个孤对电子垂直于平面。。
其他构型:包括三氮吡啶空位、吸附氮等,需通过分子动力学模拟研究其动态稳定性。
n型掺杂效应:n型掺杂效应源于氮原子比碳原子多出的一个价电子,这些额外电子会注入石墨烯的π*反键轨道,导致费米能级上移0.2-0.8 eV(具体取决于掺杂浓度),显著提高材料的本征电导率,实验测得电导率可提升2-3个数量级。
调整带隙:通过引入特定类型的氮掺杂可以精细调节带隙结构:
吡啶型氮(位于边缘或缺陷位点)会与相邻碳原子形成局域化电子态,在狄拉克点附近引入0.1-0.5 eV的带隙,这种半导体特性使其适用于场效应晶体管等器件(开关比可达104-105);
电荷重新分布:氮掺杂引发的电荷重新分布形成了独特的活性位点 – 氮原子周围电子云密度增加,而相邻碳原子因电子缺失呈现正电性,这种极化效应不仅改变了材料的功函数,还显著提升了催化活性。
氮掺杂通过增强电荷转移和调节吸附位点间距,提升其储钠容量。
DOI:10.1016/j.carbon.2018.08.071
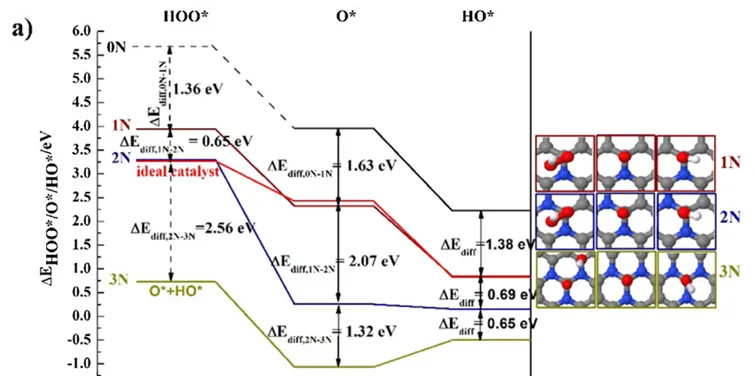
掺杂石墨烯通过调控氮原子的浓度和分布显著影响其催化性能, DFT计算表明,随着活性位点周围N浓度的增加(1N、2N、3N分别对应33%、67%、100%),O的吸附能比HO/HOO下降更快(5 eV vs. 2.7 eV),这是由于C-O键级从单键逐步转变为双键,而HO/HOO则更接近单键并遵循八电子规则。
当N原子与活性位点的距离增加(如间隔1C、2C、3C位点)时,N对HO/HOO的吸附影响迅速减弱,而O仍能感知远端N的电子效应。
此外,O作为共吸附物时对中间体吸附能的削弱作用比HO更显著(2 eV vs. 1 eV),导致理论过电位随N浓度升高(3N > 2N > 1N),但在N原子分散或共吸附HO时过电位降低。
这些结果揭示了N掺杂石墨烯的ORR活性不仅依赖于N的局部浓度和位置,还受中间体电荷再分配机制的调控,为设计高效催化剂提供了原子级理论依据。
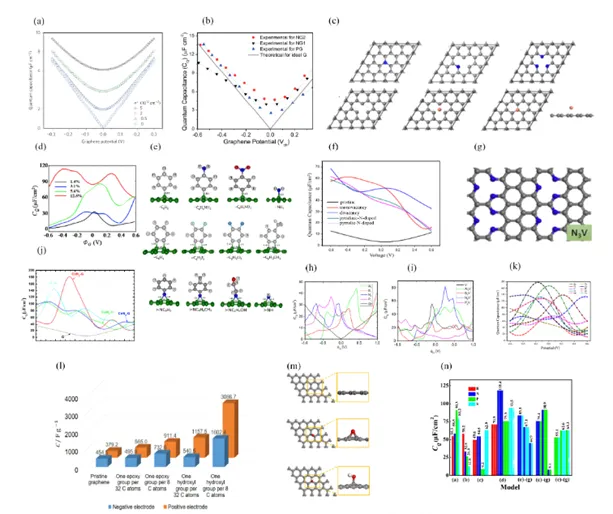
在N掺杂石墨烯的电容性能调控中,不同氮掺杂类型对总电容的贡献机制存在显著差异。
吡啶型氮(Pyridinic N)和石墨型氮(Graphitic N)通过两种互补途径协同提升电容性能:
一方面,吡啶型氮在石墨烯边缘或缺陷位点引入孤对电子,显著增加电极表面的局部电荷密度;
另一方面,石墨型氮通过取代碳原子直接向石墨烯π共轭体系注入离域电子,整体提升材料的态密度(DOS)近费米能级。
这种协同效应使得量子电容(C_Q)和电双层电容(C_EDL)同步增强。
DOI: 10.3390/nano13131932
益于理论计算化学的快速发展,计算模拟在纳米材料研究中的运用日益广泛而深入。科研领域已经逐步形成了“精准制备-理论模拟-先进表征”的研究模式,而正是这种实验和计算模拟的联合佐证,更加增添了论文的可靠性和严谨性,往往能够得到更广泛的认可。
华算科技是专业的理论计算与科研测试解决方案服务商,为高校和企业的科研团队提供材料、催化、能源、生物等领域的理论计算和测试表征解决方案。
华算科技已向国内外1000多家高校/科研单位提供了超过50000项理论计算和测试表征服务,部分计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。
添加下方微信好友,立即咨询计算服务:电话/微信:13129551561
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!