

氢析出反应(HER)是电解水制氢的核心反应,其效率直接影响绿氢技术的经济性。本文通过密度泛函理论(DFT)计算,深度解析HER反应路径、活性描述符与催化剂设计原则,为高效催化剂开发提供原子尺度洞察!
一、HER反应机理与关键步骤
HER在酸性/碱性环境中的反应路径不同,以酸性条件为例:
1.Volmer步骤:H⁺ + e⁻→ H*(氢原子吸附,ΔG_H*为关键参数)
2.Tafel或Heyrovsky步骤:
Tafel路径:2H* → H₂(g)
Heyrovsky路径:H* + H⁺ + e⁻→ H₂(g)
决速步判定:
若ΔG_H* ≈ 0 eV,反应按Tafel路径快速进行(如Pt)。
若ΔG_H*过正或过负,Heyrovsky路径主导(如MoS₂)。

二、DFT计算HER的核心参数
1. 氢吸附自由能(ΔG_H)*
· 定义:H吸附态与气相H₂的自由能差,ΔG_H = ΔE_H* + ΔZPE – TΔS。
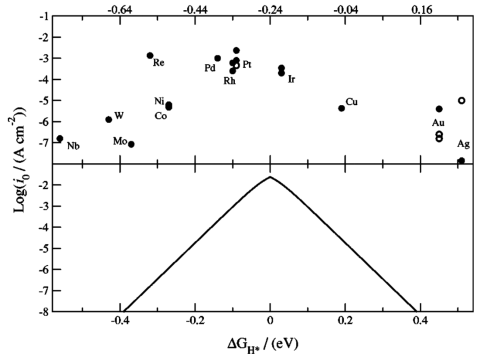
· 理想值:ΔG_H* ≈ 0 eV(火山图顶点,对应最优催化剂)。
2. 活性火山图(Volcano Plot)
· 横坐标:ΔG_H*(或描述符如d带中心ε_d)。
· 纵坐标:交换电流密度(log(j₀))或过电位(η)。
· 经典案例:Pt位于火山图顶点,ΔG_H* ≈ -0.09 eV。
3. 过电位(η)估算
· 公式:η = |ΔG_H*| / e(简化模型,e为电子电荷)。
· 精确计算:需结合活化能及微观动力学模拟。

三、DFT计算流程与关键设置
1. 模型构建
· 表面模型:金属(如Pt(111))或二维材料(如MoS₂),优化晶胞参数。
· 吸附位点:测试顶位(Top)、桥位(Bridge)、空位(Hollow)的H吸附构型。
2. 计算参数
· 泛函选择:GGA-PBE(常规体系)或RPBE(改进吸附能精度)。
· k点密度:表面模型建议≥3×3×1,真空层≥15 Å。
· 溶剂化校正:使用VASPsol或显式水层(4层H₂O分子)。
3. 自由能校正
· 零点能(ZPE):通过频率计算获取(IBRION=5 + NFREE=2)。
· 熵变(TΔS):气态H₂取实验值(0.41 eV,298 K),吸附态H*近似为0。
四、催化剂设计策略
1. 单原子催化剂(SACs):
孤立金属原子(如Pt₁/石墨烯)通过配位调控ΔG_H*。
2. 二维材料功能化:
边缘硫空位激活MoS₂基面活性(ΔG_H*从2.1 eV降至0.08 eV)。
3. 应力工程:
压缩应变降低Pt的ε_d,减弱H吸附(如Pt核@Pd壳结构)。
4. 合金化:
PtNi合金优化H吸附强度,降低Pt用量。

五、常见问题与解决方案
1. H吸附能异常:
检查自旋极化设置(H*吸附通常无需自旋极化)。
确认表面模型未发生重构(如Pt(111)未转变为Pt(100))。
2. 溶剂化效应忽略:
使用隐式溶剂模型修正ΔG_H*(误差可降低0.1-0.2 eV)。
3. 振动频率虚频:
重新优化结构或检查初始构型合理性。
找华算做计算?专业靠谱省心又省时!
益于理论计算化学的快速发展,计算模拟在纳米材料研究中的运用日益广泛而深入。科研领域已经逐步形成了“精准制备-理论模拟-先进表征”的研究模式,而正是这种实验和计算模拟的联合佐证,更加增添了论文的可靠性和严谨性,往往能够得到更广泛的认可。
华算科技已向国内外1000多家高校/科研单位提供了超过50000项理论计算和测试表征服务,部分计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。
