

一、什么是分子动力学(MD)?
分子动力学是一种基于牛顿运动方程的计算模拟方法,通过对原子间相互作用力的求解,实时追踪体系中每个原子的位移、速度和能量变化,从而揭示材料、化学反应、生命体系等在飞秒至纳秒尺度上的演化过程。它能够捕捉实验难以观测的瞬时结构、扩散路径和反应机理,已成为材料设计、药物研发、催化机理等领域的“数字显微镜”。


DOI:10.1002/wcms.1402
二、为什么MD如此重要?
- 可视化微观过程:不再是静态的结构猜测,而是动态地看到分子如何结合、离子如何迁移、蛋白如何变构。
- 洞察细节:提供实验无法捕捉的瞬时状态和微观机理,为论文提供强有力的理论支撑。
- 预测性能:在材料合成前,先通过模拟评估其稳定性、扩散能力、催化活性等关键指标,显著降低研发成本。
- 跨学科适用:从气-液界面、固-气/固-液多相体系,到电解质溶液、金属、离子液体的均相体系,再到蛋白-DNA-膜等生物大分子,MD都能提供统一的计算框架。
三、为什么选择华算科技?
在科研服务领域,版权合规与数据安全是至关重要的。而专业能力与服务品质则决定高度。华算科技凭借五大核心竞争力,成为科研工作者的信赖之选。
⭕核心优势
▶ 权威资质认证,实力获国家级认可
自成立以来,华算科技凭借深厚的技术积累与突出的创新能力,接连斩获“国家高新技术企业””创新型中小企业””科技型中小企业””专精特新企业”“CNAS”“CMA”等国家级权威资质,铸就兼具创新力与专业化的核心竞争力!未来,华算科技将继续以创新驱动发展,以专精赋能升级,为高质量发展注入强劲动力,携手合作伙伴共启价值新篇!

▶ 多学科高层次人才领衔,50000+成功服务案例
由多位深圳市高层次人才领衔,累计为全球10000+高校/企业用户提供科研服务,覆盖超1000+国内外高校、科研院所及企事业单位等,成功交付项目50000+项,以专业服务为高水平研究提供坚实支撑。深受客户信赖!

▶正版计算软件,全面保障数据合格
华算科技全面配备VASP、Materials Studio、Gaussian、MedeA等主流计算软件正版授权,从源头确保数据合规与流程可靠。12小时内快速反馈项目费用,并与客户签署严格保密协议,全方位守护客户数据安全与知识产权,让科研协作兼具高效与安心。
四、华算科技分子动力学方案:全体系覆盖,多尺度融合

华算科技分子动力学模拟服务涵盖了从多相体系到生物大分子、从吸附体系到扩散体系的全方位场景。无论是复杂的气液界面、固气界面、固液界面,还是电解质溶液、金属、离子液体等均相体系,且 Gromacs、LAMMPS、Materials Studio、CP2K 等多款业内领先的计算软件,每一款软件都具备独特的优势和适用场景 。这些计算软件相互补充,构成了华算科技强大的计算能力基础。

华算科技的专业团队能够根据不同的研究需求和体系特点,精准选择最合适的计算软件,并灵活运用其功能,为客户提供最优质的分子动力学模拟服务 。
| 业务方向 | 典型体系 | 主流软件/技术 |
| 多相体系 | 气-液、固-气、固-液界面 | Gromacs、LAMMPS、 Materials Studio |
| 均相体系 | 电解质溶液、金属、离子液体 | CP2K、VASP |
| 生物大分子 | 蛋白-DNA-膜、分子对接、QM/MM、粗粒化 | Gromacs、Gaussian、AMBER |
| 吸附/孔材料 | 多孔材料、岩土、分离膜、胶体 | LAMMPS、VASP |
| 扩散与界面 | 气体/液体/金属扩散、界面扩散 | LAMMPS、Gromacs |
| AIMD(从头算) | 非晶结构、离子迁移、化学反应 | CP2K、VASP |
五、典型成功案例(精选)
- 力学性能预测与缺陷演化
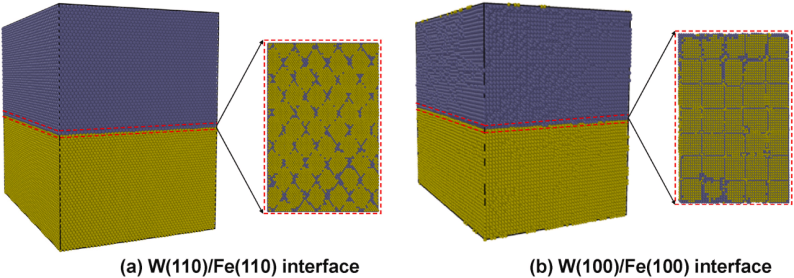
在W/Fe界面的研究中,研究者构建了W(110)/Fe(110)和W(100)/Fe(100)两种半共格界面模型,采用分子动力学模拟的方法,通过LAMMPS软件包和自行开发并修正的Fe-W嵌入原子法(EAM)势,结合ZBL势来准确预测级联效应,对W/Fe半共格界面在辐照下的缺陷演化和力学性能进行了系统研究。
模拟了在辐照过程中纯钨、纯铁以及两种半共格界面的位错环演化和应力–应变曲线,分析了不同界面取向对辐照抗性的影响,为W-Fe系统在聚变环境下的设计提供了指导。
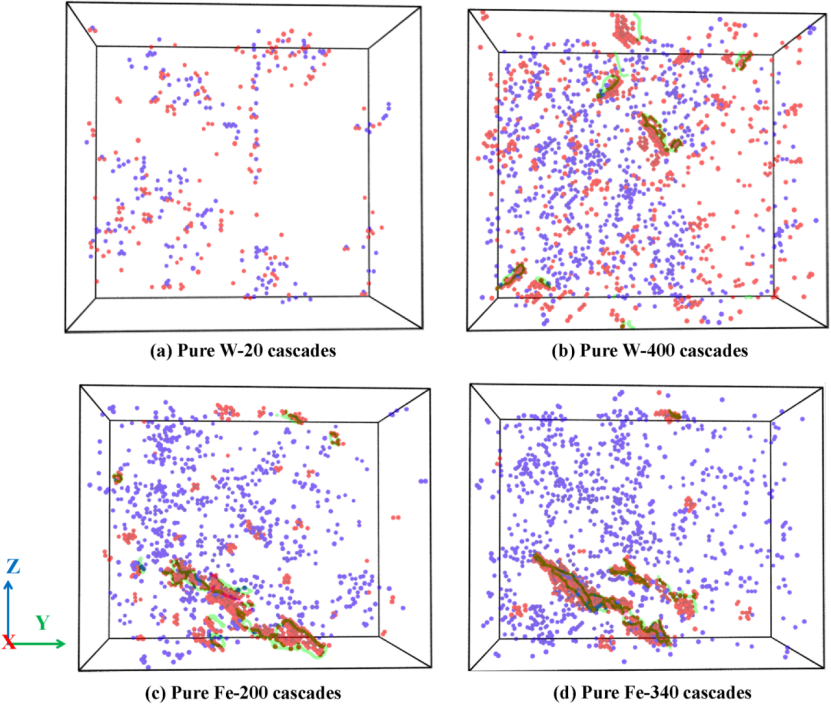
图像解析:下图展示了纯钨和纯铁在不同损伤阶段的点缺陷分布和形成的位错环。图中蓝色和红色球分别代表空位和间隙原子,绿色和粉色线分别表示在辐照下形成的柏氏矢量为1/2和的位错环。从图中可以看出,随着辐照次数的增加,纯钨和纯铁中的间隙原子不断积累,最终导致位错环的形核。在纯铁中几乎只发生位错环,而在纯钨中则只观察到柏氏矢量为1/2的位错环,这与实验观察结果一致。这表明辐照导致的损伤积累会引发位错环的形成和扩展,进而影响材料的力学性能。
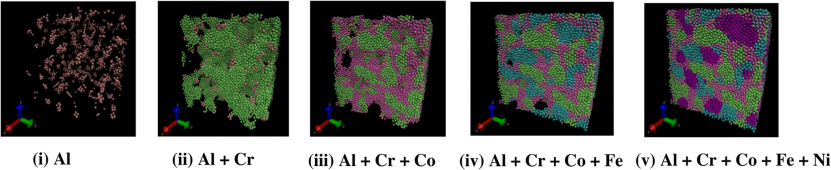
2. 纳米材料设计与性能优化
AlCrFeCuNi高熵合金的拉伸模拟展示了弹性–塑性转变的动态过程。
在Al含量为10%时,模拟轨迹显示初始阶段原子规则排列(弹性区),随着应变增加,孪晶(绿色原子簇)和层错(红色原子带)逐渐形成(屈服点),最终导致非均匀塑性变形。
通过对比不同Al含量的应力–应变曲线,发现杨氏模量随Al含量增加呈线性下降,这一结果与X射线衍射测得的晶格常数变化高度吻合。
六、选择华算,就是选择专业与信任
从单分子反应到复杂体系演化,从基础研究到产业应用,华算科技始终以“做计算,找华算”为使命,以500+博士团队护航,以50000+成功案例为证,为每一位合作伙伴提供:
✅ 定制化方案设计——精准匹配研究目标与计算方法
✅ 全流程技术支持——从模型搭建到数据分析全程跟进
✅ 高性价比服务——灵活响应需求,降低科研成本
