

Q1:朱老师,CONTCAR都是分数坐标吗?不能输出的是迪卡坐标?那优化后的晶面模型,如果要吸附分子,那要将CONTCAR再变成迪卡坐标,F和T,要重新输入?
A:vasta在输出POSCAR时,会让选择是输出分数坐标,还是笛卡尔坐标的
Q2:朱老师,请问在结构优化时候,最后结果出现了这个情况,是没有收敛吗
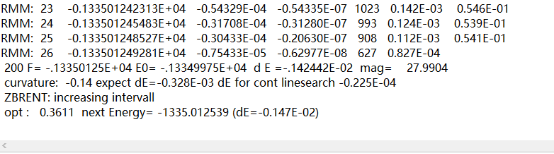

A:NSW=200,跑到设置的200步了,力没有收敛
Q3:朱老师,您好,我想问一下d轨道的pdos数据里是dxy,dyz,dxz,dx2-y2,dz2这个顺序吗?
A:前三个没问题,然后是dz2,再是x2-y2
Q4:朱老师,请问一下,像计算这种带电荷的小分子的能量应该怎么算呀?
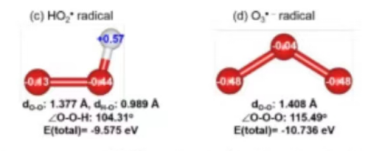
A:和算水分子一样处理
Q5:朱老师,请问一下,我算差分电荷的时候,这个反铁磁体并未加MAGMOM,整体的结构也收敛了,但是把他拆开用同样的设置计算就不收敛了,有么有什么办法能让它计算出结果可以先比对一下。
A:磁矩问题,需要设置magmom
Q6:朱老师,用vasp计算掺杂之后的结构 放到Vesta进行xrd模拟发现并没有什么变化(实验是能观察到峰偏移) 掺杂量大概6%总原子数 这是因为掺杂量不够还是因为Vesta做不到那么精细的xrd模拟?
A:是数量原因
Q7:朱老师,您好!VESTA中怎样做到坐标轴不动,晶格结构旋转平移呢?
A:不能
Q8:朱老师,请问结构优化的过程中出现这种情况怎么解决?

A:ISIF改为2试试
Q9:朱老师,用Gnuplot做图的时候想改一下横纵坐标字体的字号和大小,是什么命令啊?
A:这里不能调,要输出到图片才可以
Q10:老师,如果在输入中出现了这个问题要怎么办啊?
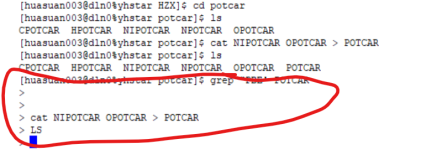
A:ctrl c
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!