

Q1:朱老师,我想请问一下计算同一物质有无磁矩的情况,得到的体系能量不同,但是磁矩都为0,是因为物质本身没有磁性吗?
A:有可能,但需要进一步查看每个原子磁矩是否都是0
Q2:朱老师,我用COHPCAR里面的pCOHp数据做图再积分 和lobster给出的ICOHP里面的数值不一样是怎么回事呢?是直接用ICOHPLIST里面的值更合理一些吗
A:因为COHPCAR是从你指定的原子对全部能量范围内积分,iCOHP是你指定的原子对从最低到0的积分,先检查一下文件给出的积分起始点是不是0,有时候不是0,低能量还有,不过没关系用他的积分就可以。用ICOHPLIST里面的值更合理
Q3:朱老师 我的COHPCAR中只有我指定的一个原子对 是怎么回事呢?
A:不会的,看看有没有写错
Q4:朱老师,我有一个问题想问您,这篇文献中提到氮化铀的实验数据是4.886,这个经过结构优化后的是4.931。请问在这个水在表面作用的时候这个数据是在单晶胞中优化的还是构建slab模型之后优化的呢?
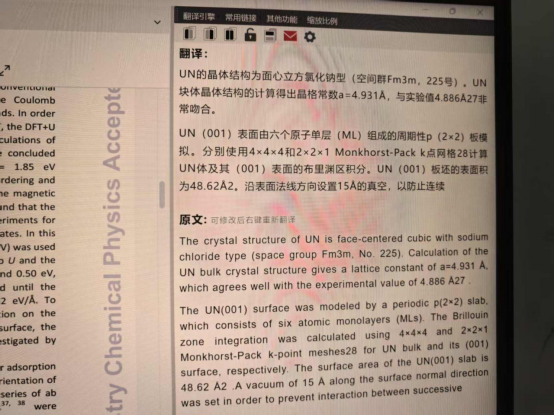

A:slab模型不需要优化晶格常数
Q5:朱老师,什么时候需要使用Monkhorst-pack K点?
A:这个和gamma差不多,精度到了都可以
Q6:朱老师,什么时候需要P1对称性去做计算?
A:对称性一般由软件自行判断,不用特意设置
Q7:二维材料计算和磁性计算需要ISYM=-1关闭对称性吗?
A:严格来说需要,但粗糙点也可以不关闭,用ISYM=0也行
Q8:朱老师,我在算态密度的时候计算完成了没有PROCAR文件输出是什么原因啊,这是计算的INCAR文件?
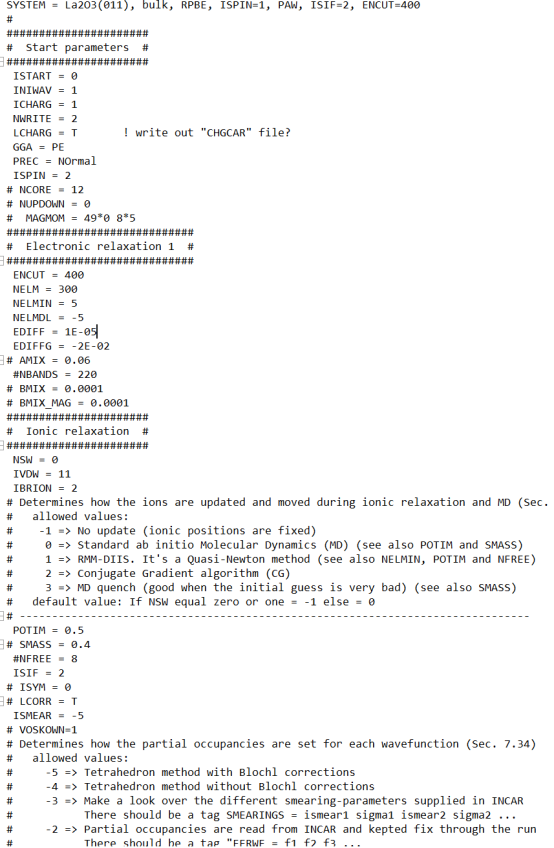

A:这个没问题,检查下VASP版本,一般5.4.4或以上才能
Q9:朱老师,请教一个问题,我用vaspsol++算AIMD,计算一段时间水分子散掉了,请问您遇到过类似问题吗?
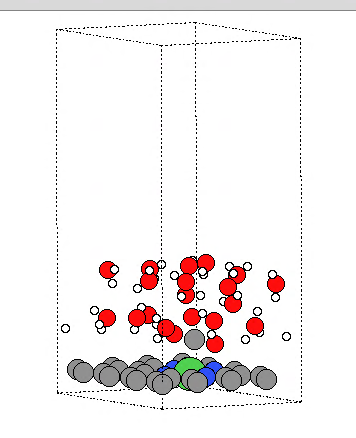
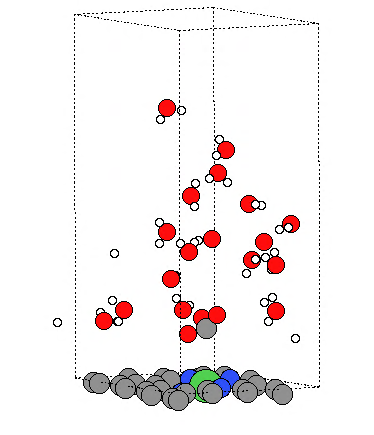
A:水可能太多了,因为质子传递是动态的水太多容易散掉,太密了会有排斥的
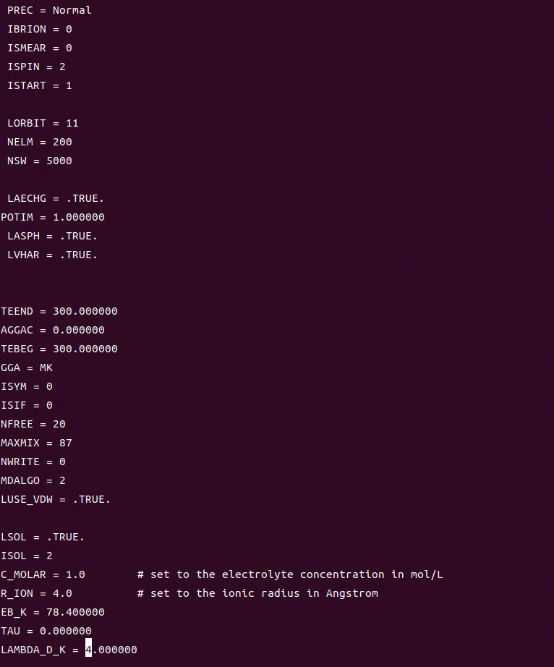
Q10:这个是什么问题,朱老师?
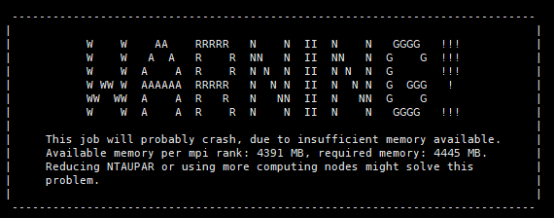
A:内存不够,增加计算资源
理论计算遇到报错?一键领取《理论计算常见错误答疑汇总》,快速排查VASP/CP2K等计算难题。