
偶极矩是描述分子内电荷分布不均匀性的关键物理量,它不仅决定了分子的极性,还深刻影响着分子间相互作用、化学反应活性以及材料的光电特性。随着计算科学的飞速发展,以密度泛函理论(DFT)为代表的量子化学计算方法已成为精确预测和深入理解分子偶极矩不可或缺的工具。
本文华算科技将从偶极矩的基本物理概念出发,系统阐述其在量子化学框架下的计算原理,并结合近年来高水平研究文献中的具体案例,特别是通过深度解析相关图表,详细探讨理论计算在揭示偶极矩物理意义、验证计算方法精度以及分析误差来源等方面的应用。
物理图像与计算理论
从物理学的角度看,偶极矩源于电荷的分离。一个最简单的物理偶极子模型由两个等量异号的点电荷+q和-q构成,它们之间的距离为d。
其电偶极矩(electric dipole moment)被定义为一个矢量 μ,其大小为q乘以d,方向由负电荷指向正电荷 。对于一个真实的分子,其电荷分布是连续的,由带正电的原子核和带负电的电子云构成。
因此,分子的偶极矩需要通过对整个体系的电荷密度分布函数ρ(r)进行积分来获得,其数学表达式为 μ = ∫rρ(r)d³r,其中r是从坐标原点指向电荷微元d³r的位置矢量。这个矢量不仅包含了偶极矩的大小(通常以德拜Debye, D为单位),还指明了分子内部正负电荷中心的相对方向,这是理解分子极性的基础。
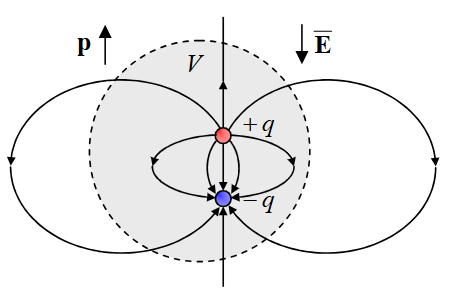

在现代量子化学计算中,求解分子偶极矩的核心任务是精确获得分子的电子波函数或电子密度。如从头算(ab initio)方法等,其计算流程通常始于对分子结构进行几何优化,找到其在势能面上的稳定构型。
随后,通过求解Kohn-Sham方程或Hartree-Fock方程,在一个迭代的自洽场(Self-Consistent Field, SCF)循环中不断优化电子密度,直至体系能量收敛至最低 。
一旦获得收敛的电子密度,偶极矩就可以作为偶极矩算符的期望值被计算出来。具体而言,计算程序会分别计算原子核贡献和电子贡献,然后将两者矢量相加得到总的分子偶极矩及其在x, y, z三个笛卡尔坐标轴上的分量 。
对于凝聚相体系,例如模拟液态水,为了更准确地描述特定分子在环境中的偶极矩,还可以采用最大局域化Wannier函数(MLWF)等高级分析方法来剖析电荷分布 。
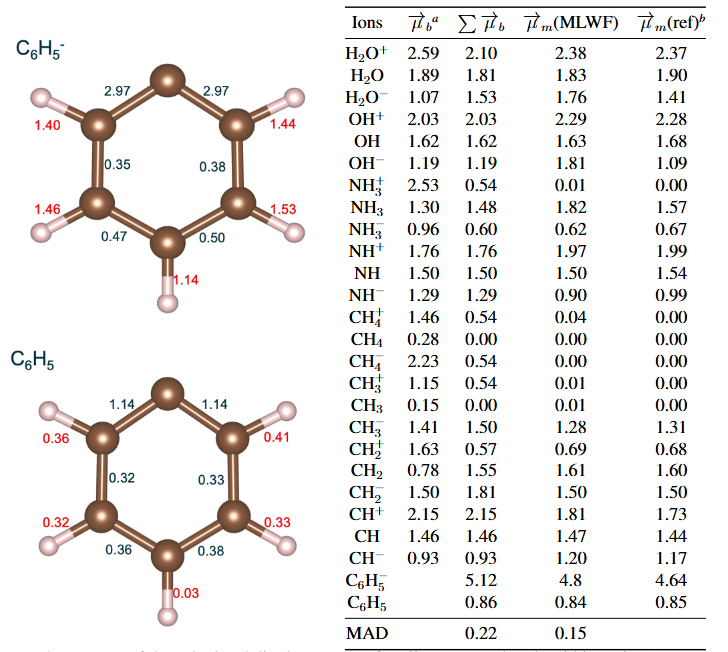
DOI: 10.1021/acs.jpclett.1c03476
理论计算文献实例
理论计算不仅能给出偶极矩的数值,更重要的是,它能将这一抽象的物理量与分子的具体结构、化学环境和动态过程联系起来,并通过可视化的方式提供直观的物理图像。
DOI:10.1088/1742-6596/2587/1/012045
分子电子结构与偶极矩的可视化分析
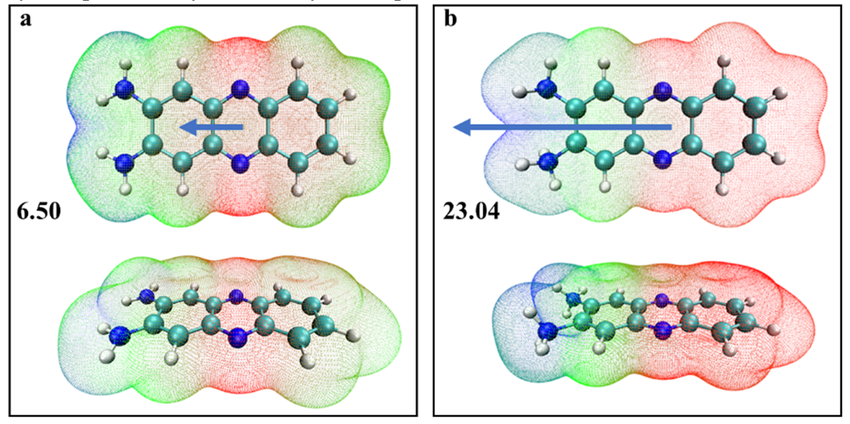
量子化学计算生成的可视化图像是连接理论与化学直觉的桥梁。例如,通过绘制分子的电子密度等值面或分子轨道图像,可以直观地判断电荷的富集与亏损区域。有文献展示了卟啉链分子的最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)的等高线图。
在这类图像中,不同颜色或轮廓线的疏密代表了电子密度的高低。如果一个分子的HOMO主要分布在一端,而LUMO主要分布在另一端,则表明该分子在发生电子跃迁时,会伴随显著的电荷从一端到另一端的转移,这将导致激发态偶极矩相对于基态发生巨大变化,这种性质对于设计非线性光学材料至关重要。
DOI:10.1039/D2TA06084A
更为直接的可视化方式是直接在分子结构上标出偶极矩矢量。中的图片就是一个极佳的例子,它展示了尼罗纳法林(Nile Red analogue)分子的诱导偶极矩。该图清晰地在分子骨架上绘制了一个红色矢量箭头,该箭头的方向和长度直观地表示了诱导偶极矩的方向与相对大小。
这个箭头从分子的富电子区域(给电子基团)指向缺电子的硝基(NO2)区域(吸电子基团),形象地揭示了分子内部由于取代基效应导致的永久性电荷分离方向。
这种可视化不仅确认了分子的极性特征,还为理解其作为溶剂探针的颜色变化机理(溶致变色效应)提供了电子结构层面的依据,因为溶剂的极性会与该分子偶极矩相互作用,从而影响其能级和吸收光谱。
动态过程与分子间的相互作用
偶极矩并非一成不变的静态参数,它在化学反应、分子振动和分子间相互作用过程中会发生动态变化。有文献表明当反应体系从初始状态向第一个过渡态演变时,偶极矩值显著上升。
这深刻地揭示了在过渡态结构中,电荷发生了剧烈的重新分布,形成了比反应物极性更强的状态。偶极矩的这一变化趋势是理解反应机理、溶剂效应以及催化剂如何通过稳定高极性过渡态来加速反应的关键信息。
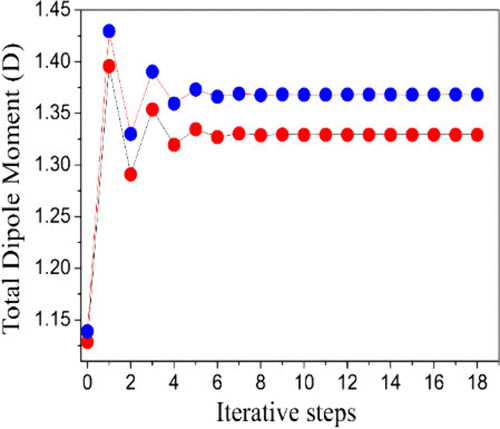
DOI: 10.1021/acsomega.4c06466
在分子间相互作用层面,偶极矩扮演着核心角色。例如,在水的模拟中,单个水分子的偶极矩是理解水作为优良溶剂的根本。研究讨论了溶剂化电子在水簇中的形成过程,指出电子是被水分子集体偶极矩所产生的静电势阱捕获并稳定的。
水分子的取向会进行调整,使其偶极矩的正端(氢原子)朝向电子,从而最大化静电吸引作用。此外,在研究蛋白质光循环时提到,蛋白质内部的电荷重排可以通过偶极矩信号的变化来追踪,其中质子转移和周围水分子的重新定向都会对总偶极矩产生贡献,这使得偶极矩的测量和计算成为探索生物大分子功能的有力工具。
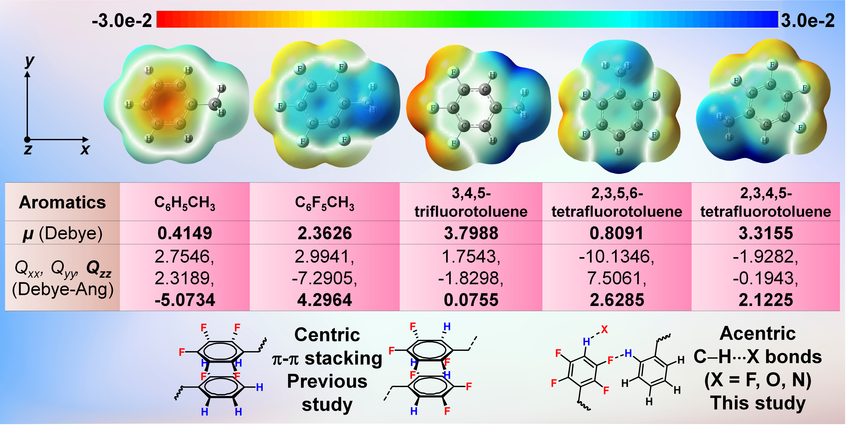
DOI:10.1002/cplu.202400320
计算方法与精度验证
理论计算的可靠性取决于所用方法的精度。因此,将不同计算方法的结果与高精度实验值或更高级别的理论计算结果进行比较,是计算化学研究中的常规操作。有学者通过散点图的形式,直观地比较了两种不同方法计算的分子偶极矩。
其横坐标是基于高级别量子力学(QM)直接计算的结果,这通常被视为“黄金标准”或参考基准;纵坐标是基于相对简化且计算成本更低的RESP(Restrained Electrostatic Potential)点电荷模型计算的结果。
图中大量的分子数据点紧密地分布在y=x的对角线周围,并且线性拟合的相关系数R²值在气相和水相中都非常接近1.0。这有力地证明了RESP模型能够在很大程度上复现高精度QM计算的偶极矩,从而验证了其在构建分子力场等应用中的合理性和可靠性。
DOI: 10.48550/arXiv.2504.16704
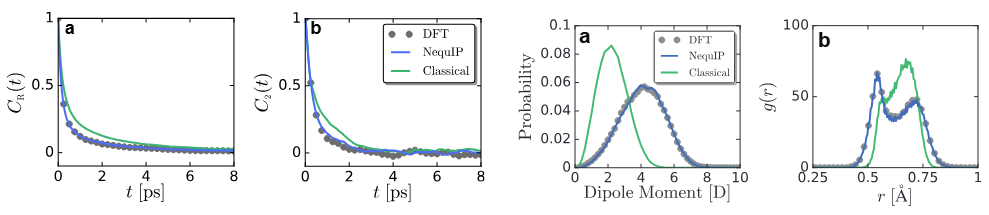
类似的比较也用于评估新兴的计算范式。对比了传统密度泛函理论(DFT)、新兴的机器学习波函数(MLP)模型以及经典力场(FF)在描述碘离子周围水分子偶极矩分布上的表现。
研究指出,经典的力场模型由于无法描述电子极化效应,其预测的偶极矩分布范围远窄于DFT和MLP模型,后者能够更好地捕捉由于结构波动引起的瞬时极化现象。这种对比分析不仅凸显了高级方法的优势,也指明了经典模型的局限性和未来改进的方向。
计算精度、收敛性与误差分析
尽管理论计算功能强大,但其结果的准确性受到多种因素的制约,主要包括所选的交换相关泛函、基组的大小以及计算模型本身。
泛函与基组对计算精度的影响
在DFT计算中,交换相关泛函的选择是决定计算精度的命脉。不同的泛函(如LDA, GGA, meta-GGA, Hybrid, Double-hybrid)对电子间相互作用的近似方式不同,因此对电荷分布的描述能力也各异。
大量的基准测试研究,通过在一个包含200个分子的偶极矩基准数据库上评估88种不同泛函的性能,系统地揭示了它们的优劣。
研究普遍发现,包含了一部分精确交换成分的杂化泛函(Hybrid functionals)和双杂化泛函(Double-hybrid functionals)通常能提供最准确的偶极矩预测,其误差可以控制在几个百分点之内。相比之下,局域密度或广义梯度近似的泛函表现则相对较差 。
基组的完备性是另一个关键因素。基组本质上是用来构建分子轨道的一组数学函数。理论上,只有当基组趋于无穷大(即完备基组极限)时,才能获得最精确的结果。在实际计算中,必须在计算成本和精度之间取得平衡。
使用较小的基组可能会导致对电子云尾部区域描述不佳,从而影响偶极矩等依赖于电荷分布的性质。有学者研究了如何通过基于DFT的基组校正方法来加速高精度耦合簇理论(CCSD(T))计算偶极矩的基组收敛性,这是一种有效降低高精度计算成本的策略。
误差评估的系统性方法与图谱分析
对计算误差进行系统性分析是评估和发展新理论方法的基石。有提供了一个极具说明性的图谱,展示了不同DFT方法在预测氟化氢(FH)分子解离过程中偶极矩变化的性能。图中横坐标为F-H键的核间距,纵坐标为计算得到的偶极矩。一条黑色的实线代表了高精度的参考曲线。
从图中可以清晰看到,在平衡键长附近(约0.9 Å),大多数泛函的预测与参考曲线吻合得很好。然而,随着键长被拉伸,不同方法的预测出现了巨大的分歧。
一些泛函(尤其是那些存在严重自相互作用误差的泛函)在键长拉大时,错误地预测偶–极矩会持续增大并趋向一个不正确的渐进行为,而正确的物理图像是,当分子解离成中性原子时,偶极矩应趋于零。
这张图谱直观地暴露了某些理论方法在处理化学键断裂等强关联或非平衡结构时的根本性缺陷,为使用者选择合适的方法提供了重要的警示。
此外,统计误差分布图也是评估方法性能的常用工具。这些图表汇总了大量分子的计算结果,通过观察误差分布的宽度、峰值位置以及是否存在异常值(outliers),研究人员可以全面地评估一个方法的普适性、稳健性和系统性偏差。
例如,一个理想的方法其误差分布应该是一个以零为中心、方差很小的正态分布。这些精细的误差分析工作,结合如差分电荷密度法和嵌入方法等,旨在消除特定误差来源的先进算法,共同推动着量子化学计算向着更高精度和更广应用范围不断迈进。
总结
综上所述,偶极矩作为一个连接微观电荷分布与宏观物质性质的核心概念,在化学、物理和材料科学中具有至关重要的理论与实践意义。
现代量子化学计算,特别是密度泛函理论,为我们提供了一套强大的工具集,不仅能够定量预测分子的偶极矩,还能通过可视化分析和动态过程模拟,赋予这一物理量生动的化学内涵。
通过对文献中各类图表的深入解读,我们看到理论计算如何在验证方法可靠性、揭示反应机理、理解分子间相互作用以及指导新材料设计等方面发挥着不可替代的作用。
然而,研究者也必须清醒地认识到计算方法固有的局限性,通过严谨的收敛性测试、系统的误差分析和与高精度基准的对比,才能确保计算结果的可靠性,并推动理论计算方法自身的持续发展。