本文系统介绍了差分电荷密度的基本概念、计算原理及其在化学和材料科学中的重要意义。差分电荷密度通过比较不同体系的电荷密度差异,直观地揭示了电子在化学反应或材料相互作用中的重新分布情况。
文章详细阐述了差分电荷密度在化学键形成、电子转移方向分析以及材料性能研究中的关键作用,并介绍了三维可视化和二维切面分析等常用可视化方法。研究表明,差分电荷密度不仅能够帮助理解共价键、离子键等化学键的本质,还能为材料设计、催化机制研究等提供重要理论依据。
通过分析电子在空间中的分布变化,差分电荷密度为深入探索材料电子结构和反应机理提供了强有力的工具。
差分电荷密度定义与原理
差分电荷密度是两个体系(如成键前后、吸附前后等)的电荷密度之差,其数学表达式为:△ρ= ρAB – ρA– ρB。
其中,ρAB是体系AB的电荷密度,它描述了体系AB整体的电子云分布特征。ρA
和ρB分别是片段A和B的电荷密度。 通过计算△ρ,可以得到体系AB相对于片段A和B单独存在时电荷密度的变化情况。
差分电荷密度图通过颜色变化直观地表示电荷密度的增减,通常在可视化时,红色表示电子增加,意味着该区域电子云密度相对片段A和B单独存在时有所增强;蓝色表示电子减少,即该区域电子云密度相对降低 。
这种通过颜色映射电荷密度变化的方式,使得我们能够直观地观察到电子在不同体系或结构之间的重新分布情况。

物理意义
差分电荷密度能够直观反映电子在不同体系或结构之间重新分布情况。在化学反应中,当原子相互靠近形成化学键时,电子会在原子间重新分布,通过差分电荷密度分析,可以清晰地看到电子从哪些原子转移到哪些原子,以及电子在成键区域的聚集或离域情况。
在理解化学键形成方面,差分电荷密度起着关键作用。对于共价键,在成键过程中电子云会在两个原子之间聚集,形成共享电子对,此时差分电荷密度图会在成键区域显示出电子增加的区域(如黄色),表明电子在该区域的富集,这是共价键形成的重要特征。
对于离子键,电子会从电负性较小的原子完全转移到电负性较大的原子,差分电荷密度图能清晰地展示这种电子的完全转移,电负性较小原子周围显示电子减少(如蓝色),电负性较大原子周围显示电子增加(红色),从而帮助我们深入理解离子键的本质是电子的得失和静电相互作用。
在研究电子转移方向上,差分电荷密度提供了直接的信息。在氧化还原反应中,电子从还原剂转移到氧化剂,通过分析差分电荷密度,可以准确地确定电子转移的起点和终点,量化电子转移的程度。
在半导体材料中,当存在杂质或缺陷时,电子会在杂质原子或缺陷周围重新分布,差分电荷密度能够揭示这种电子重新分布的情况,从而帮助我们理解杂质或缺陷对半导体电学性能的影响。
可视化方法
三维可视化
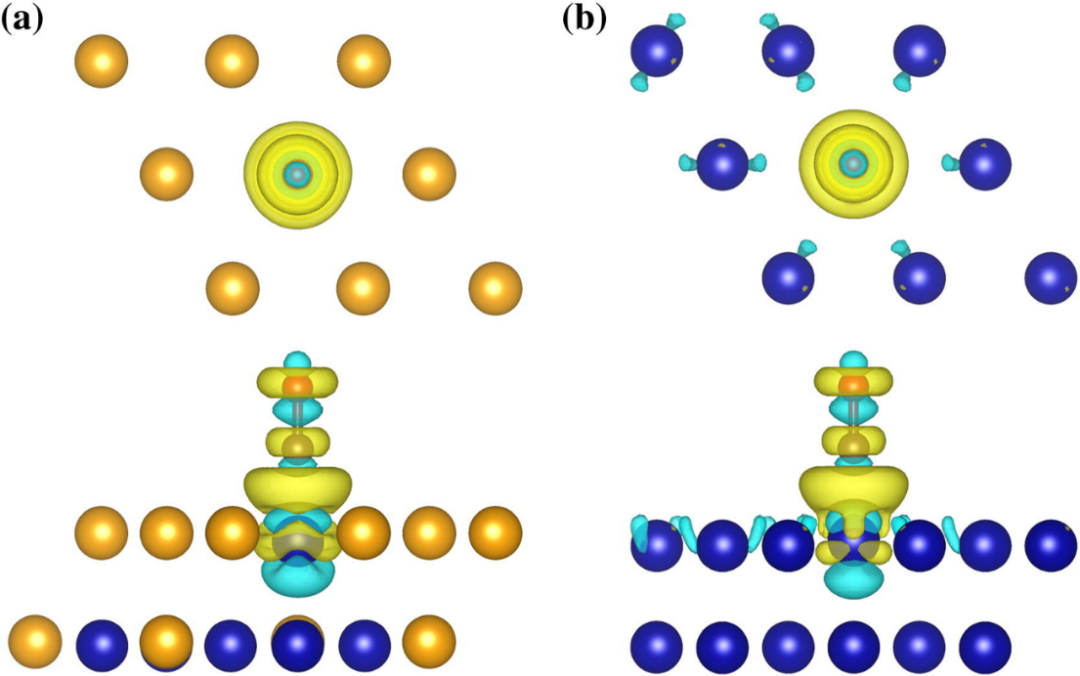
使用VESTA软件可以生成三维差分电荷密度图,通过颜色变化直观地观察电子的重新分布。以CO分子与金属表面的相互作用为例,在生成的三维差分电荷密度图中,从金属原子区域到CO分子区域,电子密度的变化通过颜色的过渡清晰地展现出来 。
金属原子周围蓝色区域表示电子减少,这是因为电子从金属原子流出;而CO分子中碳原子和氧原子周围的黄色区域表示电子增加,表明电子流向了CO分子 。通过旋转三维视图,可以从不同角度全面观察电子在金属与CO分子之间的转移路径和分布情况,为深入理解CO分子与金属表面相互作用的微观机制提供直观依据 。
二维切面分析
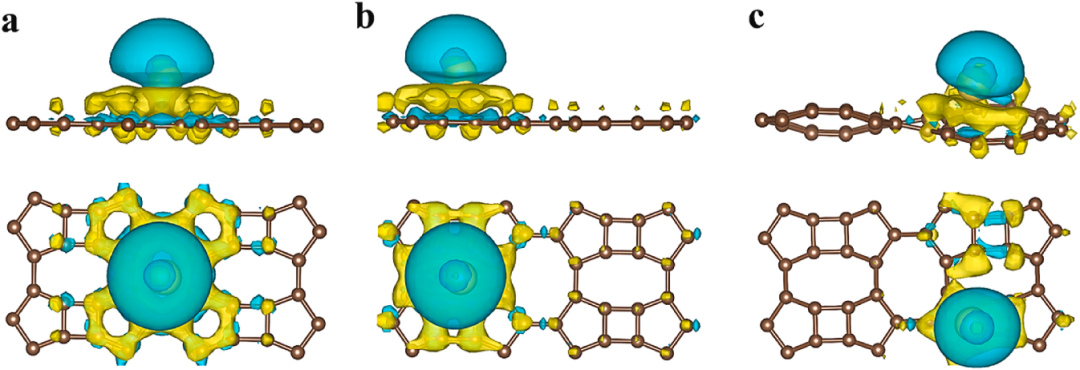
通过选择特定平面,可以VESTA生成二维差分电荷密度图。差分电荷密度(CDD)分析揭示了Ti修饰的SVBP-Ti₂和SVBPAI-Ti₂系统在吸附20个H₂分子后的电荷重新分布机制。
通过等值面可视化,图中蓝色区域(电荷积累)和黄色区域(电荷耗尽)清晰地展示了H₂分子与Ti原子之间的电子转移行为。在SVBP-Ti₂系统中,Ti的3d轨道与H₂的σ轨道发生显著杂化,导致电子从Ti向H₂的σ*反键轨道转移,形成极化共价键,从而弱化了H-H键(键长从0.74 Å增至0.81 Å)。
SVBPAI-Ti₂系统则进一步通过Al掺杂增强了电荷离域性,使Ti-d轨道与基底(B-p、P-p、Al-p)的杂化更显著,从而提升了H₂吸附的稳定性。
平面平均差分电荷密度
使用平面平均差分电荷密度分布可以更清晰地观察电子的重新分布。差分电荷密度(DCD)分析揭示了Si-MoS₂、Ge-MoSe₂和Sn-MoTe₂与C₂H₄分子之间的电荷转移和化学键形成机制。
通过等值面设置为0.001e/ų的可视化,图中蓝色区域(电荷耗尽)和黄色区域(电荷积累)清晰地展示了C₂H₄分子与掺杂原子(Si、Ge、Sn)之间的电子共享行为。这种共享电子云的形成表明C₂H₄与掺杂原子之间形成了共价键,其中Si-C键的电子重叠最为显著,其次是Ge-C和Sn-C键,这与COHP分析中键强度的趋势一致。
DCD结果进一步验证了吸附过程中电荷的重新分布,解释了为何Si-MoS₂表现出最强的吸附能力和最高的灵敏度,同时也为理解材料电子传输性质的变化提供了微观层面的依据。
CO₂RR中差分电荷密度应用
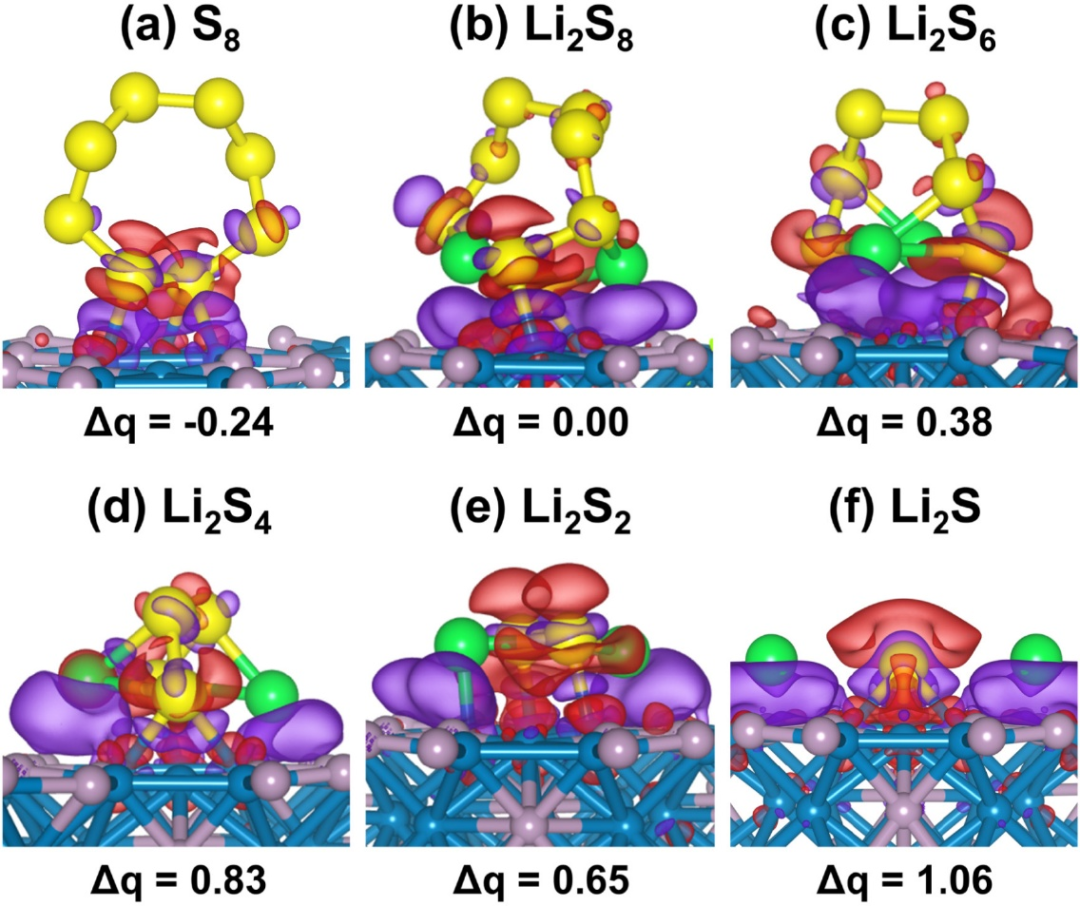
图中的电荷密度差分析在研究中具有重要的意义,它直观地揭示了锂多硫化物(LiPSs)与Ni₂P(0001)表面之间的电子相互作用机制,为理解吸附过程中的电荷转移和化学键形成提供了关键证据。
通过CDD图可以清晰地观察到吸附过程中电荷的重新分布情况,其中紫色区域表示电荷积累,而品红色区域表示电荷耗竭。这种电荷的重新分布直接反映了LiPSs与Ni₂P(0001)表面之间的电子转移方向以及化学键的强度,从而为解释吸附能的来源和吸附稳定性提供了理论依据。
具体来说,CDD分析表明,LiPSs与Ni₂P(0001)表面之间的相互作用主要通过Ni-S键的形成来实现。在吸附过程中,电子从LiPSs的硫原子向Ni₂P表面的Ni原子转移,导致电荷在Ni-S键区域积累,同时在LiPSs的硫原子周围出现电荷耗竭。这种电荷转移现象表明Ni₂P表面具有较强的极性,能够通过化学键的方式锚定LiPSs,从而有效抑制“穿梭效应”。
此外,CDD图还显示,不同LiPSs物种(如Li₂S₈、Li₂S₆、Li₂S₄、Li₂S₂和Li₂S)在Ni₂P表面上的电荷分布模式存在差异,这进一步说明了吸附强度与LiPSs分子结构之间的相关性。例如,Li₂S的吸附表现出最显著的电荷转移和电子局域化,这与其实验中观察到的最高吸附能(-5.28 eV)和自发解离行为一致。
CDD分析还揭示了Ni₂P(0001)表面三重Ni位点的活性本质。这些位点通过与硫原子的强相互作用,形成了稳定的Ni-S键,从而为LiPSs提供了高效的锚定位点。这种相互作用不仅抑制了LiPSs的溶解,还促进了硫还原反应的动力学过程。
此外,电荷密度差的变化还反映了LiPSs分子内Li-S键的弱化现象,这与实验中观察到的Li-S键伸长(如Li₂S中Li-S键伸长1.52 Å)相吻合,进一步证实了Ni₂P表面在催化LiPSs转化中的积极作用。
从更广泛的角度来看,CDD分析为Ni₂P作为锂硫电池宿主材料的性能优化提供了理论指导。通过揭示电荷转移和键合机制,研究为设计具有更高吸附能力和催化活性的电极材料指明了方向。
例如,可以通过调控Ni₂P表面的电子结构或暴露更多活性晶面来进一步增强其与LiPSs的相互作用。此外,CDD结果还与其他分析手段(如吸附能计算和态密度分析)相互印证,共同构建了对Ni₂P(0001)表面吸附行为的全面理解。
总之,CDD分析不仅为Ni₂P(0001)表面与LiPSs的相互作用提供了直观的电子层面证据,还深化了对锂硫电池中宿主材料设计原则的认识。其意义不仅限于本研究的结论,还为未来开发高性能锂硫电池电极材料奠定了理论基础。