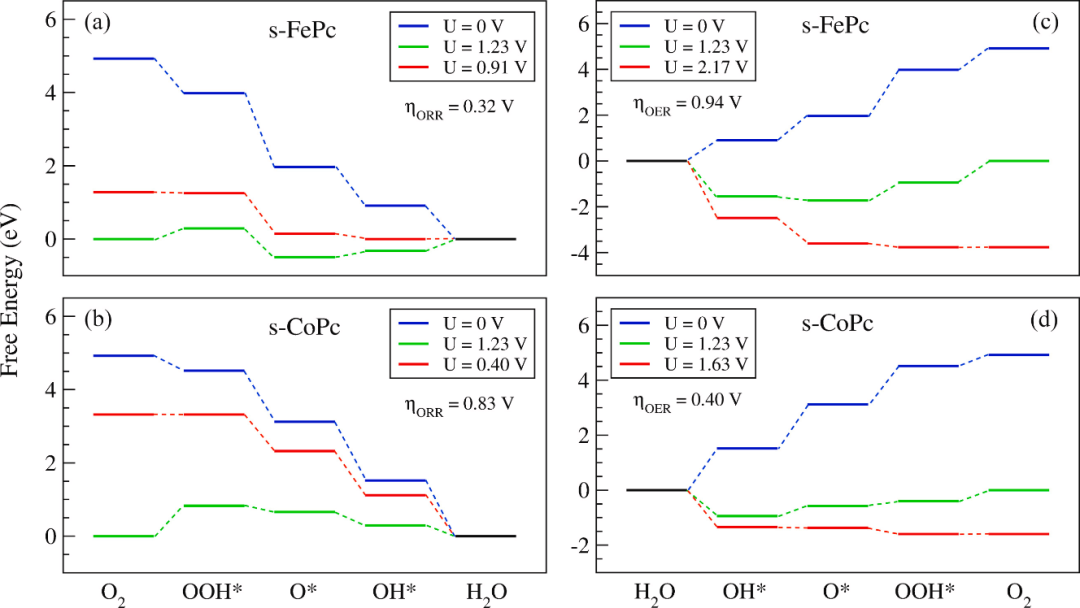
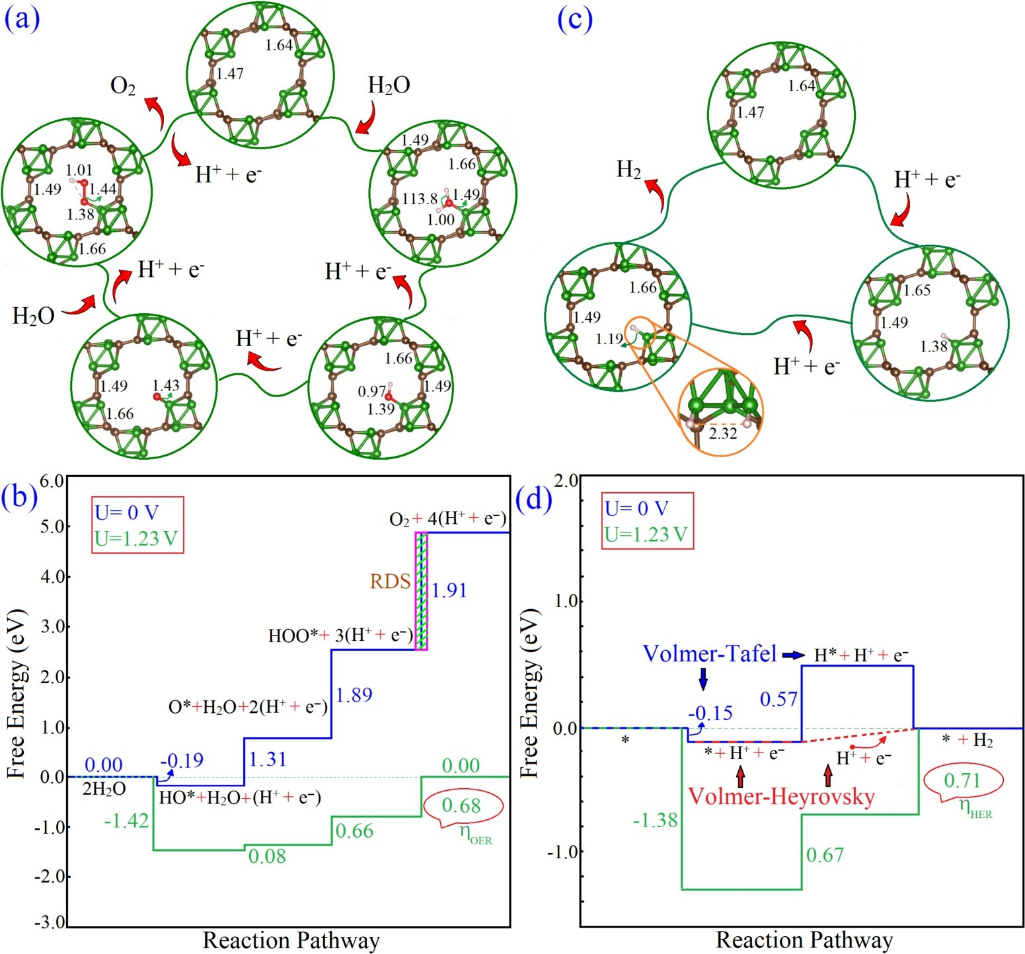
本文系统阐述了过电位的基本概念及其在电催化反应中的重要作用。过电位作为实际反应电压与理论电压的差值,反映了电化学过程中的能量损耗。文章从热力学和动力学角度分析了过电位的产生机制,包括活化能垒和电荷转移阻力等因素的影响。
通过理论模型和计算方法,探讨了过电位与反应速率、能量效率之间的关系,并阐述了其在优化催化反应中的指导意义。研究结果表明,深入理解过电位的本质对于设计高效电催化体系具有重要价值,为降低反应能耗、提高催化效率提供了理论基础。
过电位的定义
过电位是电催化反应中一个至关重要的概念,它指的是在电催化或光电催化反应过程中,达到一定电流密度时所需实际电压超过理论电压的部分。在理想的热力学平衡状态下,电催化反应所需的运行电位即为平衡状态下的电位,此时反应可以在最低的能量消耗下进行。
然而,在实际的反应体系中,由于存在多种动力学因素的阻碍,反应往往不能在理论电位下顺利发生,而是需要额外施加一定的电压,这部分超出理论值的电压就是过电位。从微观角度来看,过电位的产生主要源于电极反应过程中的活化电阻和电荷转移电阻等。
在电极表面,反应物分子需要克服一定的能量障碍才能发生化学反应,这就导致了活化电阻的存在。同时,电荷在电极与反应物之间的转移也并非完全可逆,存在一定的阻力,即电荷转移电阻。这些电阻的存在使得反应需要更高的电位来驱动,从而产生了过电位。
过电位对催化反应的动力学和热力学都有着显著的影响。从动力学角度而言,过电位直接影响着反应速率。根据Butler – Volmer方程,反应电流密度与过电位之间存在指数关系,过电位的增加会使反应电流密度迅速增大,从而加快反应速率。
在析氢反应中,较高的过电位能够促进氢离子在电极表面得到电子并转化为氢气,提高析氢反应的速率。然而,过电位的增大也意味着需要消耗更多的能量来驱动反应,这在一定程度上降低了反应的能量效率。
从热力学角度来看,过电位的存在改变了反应的吉布斯自由能变化,使得反应的实际驱动力发生改变。在一些电催化反应中,过高的过电位可能导致反应的热力学可行性降低,甚至使原本在热力学上可行的反应变得难以发生。
在二氧化碳电还原反应中,如果过电位过高,可能会使反应的能量消耗过大,同时还可能引发副反应,降低目标产物的选择性。因此,深入理解过电位的本质和影响因素,对于优化催化反应、提高反应效率和选择性具有重要的理论和实际意义。

反应自由能图构建与过电位计算
在利用DFT计算研究催化反应过电位时,构建反应自由能图是一个关键步骤。反应自由能图能够直观地展示催化反应过程中各个步骤的能量变化,为理解反应机制和计算过电位提供重要依据。
构建反应自由能图首先需要确定催化反应的具体路径和涉及的中间体。以析氧反应(OER)为例,其在酸性介质中的反应方程式为2H2O⇌ O2+4H++4e−,标准平衡电极电位E0=1.23V(vs. RHE,可逆氢电极)。该反应过程通常涉及多个电子转移步骤和不同的中间体,基本反应步骤可表示为:
第一步:∗H2O⇌∗OH+H++e−
第二步:∗OH⇌∗O+H++e−
第三步:∗O+H2O⇌∗OOH+H++e–
第四步:∗OOH⇌O2+H++e–+∗
其中,*表示催化剂表面的活性位点,∗OH、∗O、∗OOH为反应中间体。
确定反应路径和中间体后,使用DFT方法计算每个步骤的吉布斯自由能变化△G。吉布斯自由能变化的计算公式为△G=△E + △ZPE – T△S,其中△E是电子能量变化,可通过DFT计算直接得到;△ZPE是零点能修正,考虑了分子振动的基态能量;T是温度;△S是熵变。
在实际反应中,由于存在过电位,反应需要更高的电位才能发生。过电位的计算与反应自由能图密切相关。在反应自由能图中,过电位通常由反应路径中吉布斯自由能变化最大的步骤(即决速步)决定。在 OER 反应中,对于每个步骤,都推导出了详细的吉布斯自由能变化(ΔG1、△G2、△G3、△G4),从而能够计算出过电位(ηOER):ηOER=(max(ΔG1、△G2、△G3、△G4)/e)-1.23V
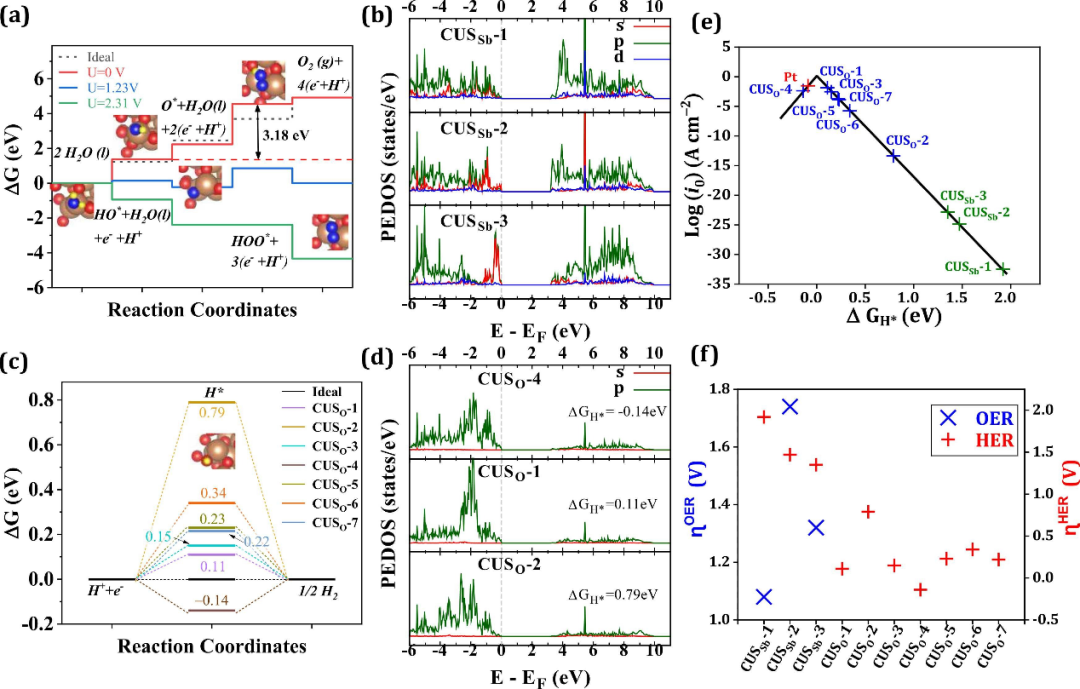
案例分析
理论计算揭示β-Sb₂O₃ (1 2 1)晶面的低过电位OER催化机制
在下图中,通过理论计算研究了理解正交相β-Sb₂O₃的(121)晶面在电催化水分解反应中的氧析出反应(OER)性能提供了关键的理论依据。通过密度泛函理论(DFT)计算,作者系统分析了该晶面的电子结构、中间体吸附行为以及反应自由能变化,并重点评估了其过电位特性,揭示了β-Sb₂O₃作为高效双功能催化剂的潜力。
首先,研究通过GGA-PBE和GGA-PBE-D3泛函计算了(121)晶面的几何结构与电子性质。结果表明,该晶面在表面存在不对称的电荷分布和配位不饱和的活性位点(CUS₅₀),这些位点由Sb³⁺离子的孤对电子和表面氧原子共同构成。
通过Bader电荷分析发现,表面Sb离子的电荷分布差异显著,尤其是CUS₅₀-1位点表现出较低的电荷转移(1.70e),使其成为OER中间体吸附的理想位点。此外,投影态密度(PDOS)分析显示,价带顶(VBM)主要由O2p轨道和Sb5p轨道杂化而成,而导带底(CBM)则以Sb5p轨道为主,这种电子结构特性为OER过程中的电荷转移提供了有利条件。
在OER机理研究中,作者通过计算H₂O分子在表面的吸附能(Eₐdₛ)验证了反应初始步骤的可行性。数据显示,H₂O在CUS₅₀-1位点的吸附能为-0.522 eV,表明其自发吸附能力。随后,通过自由能图分析了OER的四步反应路径(HO* → O* → HOO* → O₂),发现HOO*的形成(ΔG₃)是OER的决速步。
这一步骤的自由能变化(ΔG₃ = 2.314 eV)决定了整体反应的过电位。计算得到的理论过电位(ηᴼᴱᴿ)为1.08 V,显著低于许多常见金属氧化物催化剂(如TiO₂的1.19 V和WO₃的1.04–1.10 V),表明β-Sb₂O₃具有优异的OER活性。
进一步分析表明,CUS₅₀-1位点的低过电位与其独特的电子结构密切相关。该位点的Sb5p轨道在VBM中贡献显著,而Sb5s轨道参与较少,这种轨道分布优化了中间体(如O和HOO)的吸附强度。
相比之下,CUS₅₀-2和CUS₅₀-3位点因Sb 5s轨道主导或杂化不足,导致中间体吸附过强或过弱,从而表现出更高的过电位(分别为1.74 V和1.32 V)。此外,作者还通过普适标度关系验证了HO与HOO吸附能差(ΔEᴴᴼᴼ* − ΔEᴴᴼ* ≈ 3.2 eV)与高效催化剂RuO₂的相似性,进一步支持了β-Sb₂O₃的催化潜力。
为深入理解催化活性来源,研究对比了不同金属氧化物催化剂的OER过电位。结果显示,β-Sb₂O₃的过电位(1.08 V)与RuO₂(0.37 V)和IrO₂(0.56 V)虽有一定差距,但优于TiO₂(1.04–1.19 V)、WO₃(1.04–1.10 V)和Fe₂O₃(0.79–1.47 V)等材料。这一性能归因于(121)晶面的不对称几何结构和电子离域效应,其能够有效调节中间体的吸附强度,避免过强或过弱的结合,从而降低反应能垒。
此外,研究还通过表面Pourbaix图分析了不同电位和pH条件下表面吸附物种的稳定性。在OER电位范围内(U > 1.23 V vs. RHE),表面倾向于形成HO和O覆盖层,这些吸附态在酸性条件下(pH = 0)的稳定电位分别为0.43V和0.66V(vs. RHE)。这一结果与自由能计算一致,进一步证实了HOO*形成步骤的热力学限制性。
综上所述,下图的理论计算不仅揭示了β-Sb₂O₃的{1 2 1}晶面作为OER催化剂的活性位点与反应机制,还通过过电位分析明确了其性能优势。该研究为设计低成本、高效的双功能电催化剂提供了重要的理论指导,尤其强调了表面电子结构调控对优化催化活性的关键作用。未来研究可进一步探索掺杂或异质结构策略,以降低过电位并提升β-Sb₂O₃的实际应用性能。
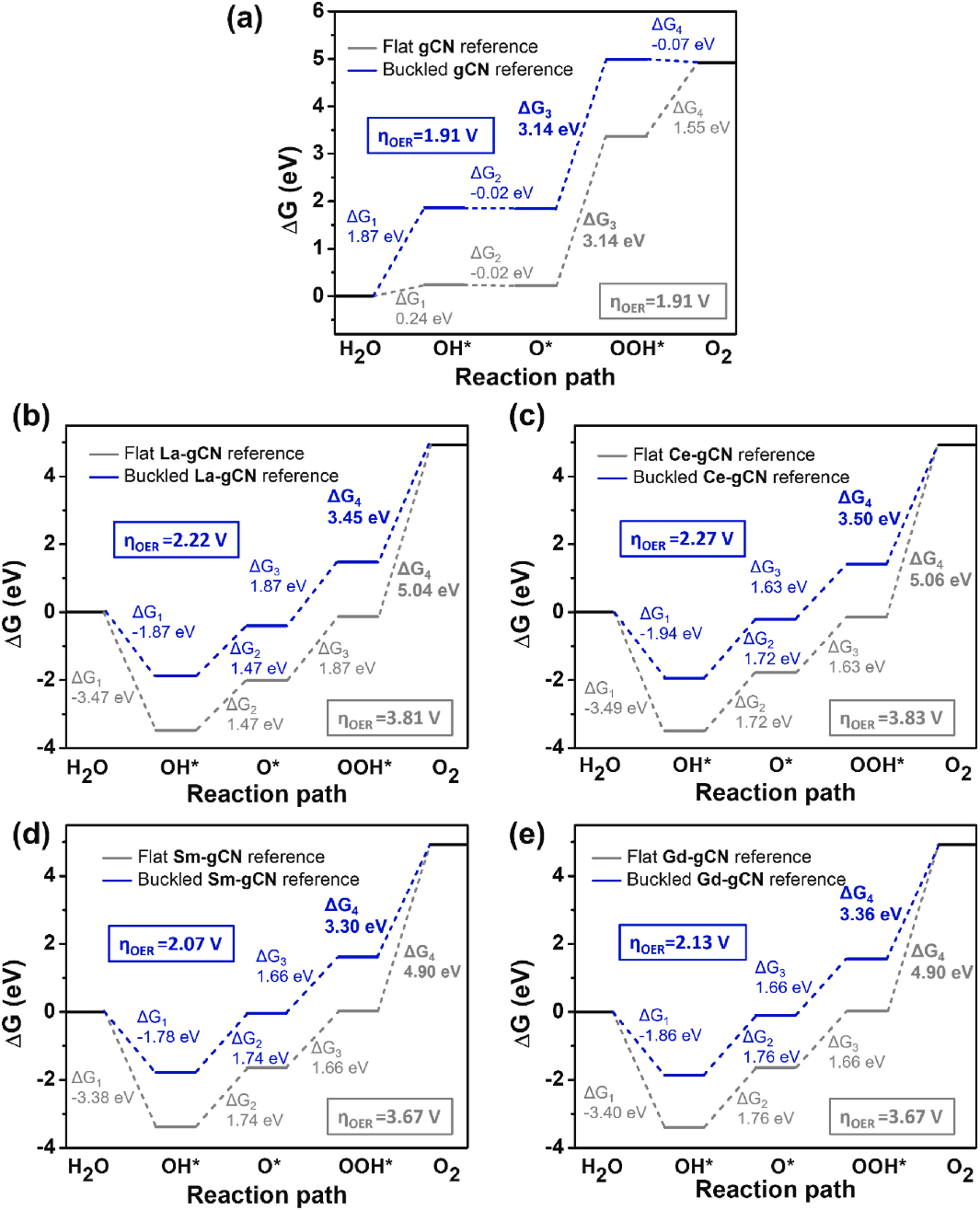
稀土单原子催化剂在析氧反应中的过电位调控机制
在这篇文献中,通过密度泛函理论(DFT)计算系统地研究了不同稀土单原子催化剂(RE-SACs)在石墨相氮化碳(g-C₃N₄)单层上的析氧反应(OER)自由能路径,重点揭示了过电位(ηOER)与催化剂电子结构及金属–载体相互作用的关联。
研究首先对比了平坦和弯曲构型的g-C₃N₄及其负载的La、Ce、Sm、Gd单原子催化剂的OER性能,发现RE单原子的引入显著改变了反应中间体(OH*、O*、OOH*)的吸附自由能(ΔG),进而影响OER的决速步骤(PDS)和过电位。
对于原始g-C₃N₄,OER的PDS为O→OOH*步骤,ΔG₃高达3.14 eV,对应过电位为1.91 V,表明其催化活性较低。而RE-SACs的引入通过强金属–载体相互作用优化了中间体吸附,使得OH*→O*→OOH*的能垒降至1.47–1.83 eV,显著低于原始g-C₃N₄。
然而,由于RE单原子对中间体的吸附过强,最终步骤OOH*→O₂成为新的PDS,导致过电位升高(2.07–2.27 V)。尽管如此,较重的Sm和Gd单原子催化剂仍表现出更低的ηOER(分别为2.07 V和2.13 V),优于较轻的La和Ce(2.22 V和2.27 V),这归因于其4f轨道的强自旋极化特性增强了与g-C₃N₄的轨道杂化,从而优化了电子转移效率。
研究进一步通过差分电荷密度(CDD)和Bader电荷分析揭示了RE单原子与中间体的电荷再分配机制。例如,OER过程中,RE原子(如Sm和Gd)始终带正电(1.83–2.05 |e|),表明电子从金属向吸附的OH、O和OOH转移,形成稳定的离子键合。这种电荷转移削弱了OOH的解吸能垒,但未能完全克服其强吸附导致的动力学限制。
此外,通过对比平坦和弯曲构型的参考体系,发现弯曲构型的RE-gCN具有更负的形成能和更高的电化学稳定性,但其OER过电位的降低幅度有限,说明构型变化对催化性能的调控作用弱于金属–载体电子相互作用的本质影响。
理论计算还结合能带结构和态密度(DOS)分析,指出RE单原子的引入使费米能级(EF)向导带底(CBM)移动,减小了带隙(如Sm-gCN带隙降至0.58–1.39 eV),并分离了电子给体(N-2p主导的价带顶)与受体(RE-4f/5d参与的导带底)轨道,抑制了电子–空穴复合。
尤其是Sm和Gd的4f轨道在EF附近表现出显著的自旋极化,为OER提供了更多可用的电子态,增强了催化活性。此外,RE-gCN的功函数(3.26–3.79 eV)较原始g-C₃N₄(4.31–5.19 eV)显著降低,进一步促进了界面电子转移,加速了OER动力学。
综上所述,理论计算不仅量化了不同RE-SACs的OER过电位,还通过电子结构分析阐明了性能差异的物理根源:较重的Sm和Gd单原子因强4f轨道耦合和优化的电荷转移能力,表现出更低的ηOER,而较轻的La和Ce则因金属–载体相互作用较弱导致催化活性受限。
尽管RE-SACs对OOH*的强吸附限制了其OER性能的进一步提升,但该研究为设计高效稀土基催化剂提供了关键的理论依据,即通过调控4f轨道活性和载体电子结构可进一步优化过电位。未来研究或需探索双原子催化或杂原子掺杂策略,以平衡中间体吸附强度,实现OER性能的突破。