

催化反应的核心始于吸附机理——分子通过物理吸附(范德华力)或化学吸附(化学键重组)“锚定”在催化剂表面,直接决定反应物的富集、活化与选择性。DFT计算作为原子尺度的“虚拟显微镜”,从四维度解析吸附本质。
结构优化揭示分子构型变化(如甲醇在金表面C-O键拉伸);吸附能计算量化结合强度(如vdW修正提升CO₂预测精度);电子结构分析追踪电荷转移(如Pd掺杂上移d带中心强化吸附);反应路径模拟定位能垒决速步(如CO→CHO是CO₂甲烷化瓶颈)。
吸附的重要作用
吸附机理在催化反应中起着至关重要的作用,主要体现在以下几个方面:
吸附是催化反应的前提:反应物必须首先吸附在催化剂表面,才能发生后续的化学反应。例如,在碳纳米管(CNTs)作为电极材料的催化反应中,反应物(如4-羟基苯酚)通过吸附作用被固定在催化剂表面,从而启动反应。此外,吸附过程还可能涉及物理吸附或化学吸附,具体取决于反应物与催化剂表面的相互作用强度。
吸附改变了反应物的性质:吸附过程中,反应物分子可能发生变形或解离,从而降低反应的活化能。例如,在金属表面,饱和烃类分子需要断裂至少一个碳-氢键才能形成表面化学吸附物种,从而启动反应。这种吸附诱导的分子变形或解离是催化反应加速的关键机制之一。
吸附影响反应路径和选择性:吸附不仅影响反应速率,还可能改变反应路径,从而影响产物的选择性。例如,在TiO₂光催化反应中,O₂、H₂O和有机物的吸附行为直接影响了反应的进行方式和产物的生成。此外,吸附分子的存在可能通过弱吸附或辅助吸附的方式影响反应中间体的形成和转化。
吸附与脱附的循环是催化反应的核心:吸附后的反应物在催化剂表面发生反应生成产物,随后产物通过脱附作用离开催化剂表面,从而实现催化循环。例如,在CO₂甲烷化反应中,吸附增强催化方法通过吸附作用提高了反应活性,并实现了产物的高效生成。此外,吸附和脱附的动态平衡对催化剂的稳定性和重复使用性至关重要。
吸附机制的多样性:吸附机制可以分为物理吸附和化学吸附,前者基于范德华力,后者涉及化学键的形成。不同的吸附机制适用于不同的反应类型和催化剂体系。例如,在氧气还原反应中,氧气分子可以通过解离机制、配位机制或过氧机制在催化剂表面吸附。
吸附机理在催化反应中不仅决定了反应是否能够发生,还深刻影响了反应的速率、选择性和效率。理解吸附机制对于设计和优化高效催化剂具有重要意义。
吸附分类
物理吸附:靠微弱的范德华力(分子间引力)结合,不破坏分子结构。例如活性炭吸附废气中的硫化氢(H₂S),分子完整附着在孔隙表面。
化学吸附:分子与催化剂形成强化学键,结构被改变。例如CO₂在镍催化剂表面断裂C=O键,生成CO和O原子。
不同吸附类似决定催化效率,主要体现在:
浓度效应:物理吸附让反应物富集在表面,碰撞概率大增。
活化效应:化学吸附弱化分子内部化学键,例如TiO₂表面吸附的水分子(H₂O)被解离成·OH自由基,氧化能力飙升。
选择性控制:手性分子(如药物中间体)在铂催化剂表面的特定吸附位点,可实现>90%的目标产物选择性。
DFT计算分析反应机理
结构优化
确定吸附时分子如何“躺”在催化剂表面,精确到键长、键角的变化。如金表面吸附醇盐分子,下图展示DFT优化的乙醇酸和乳酸在金(111)表面的吸附构型。分子的氧原子(红色)紧贴金原子(银色),O-Au距离仅3 Å和2.09Å。
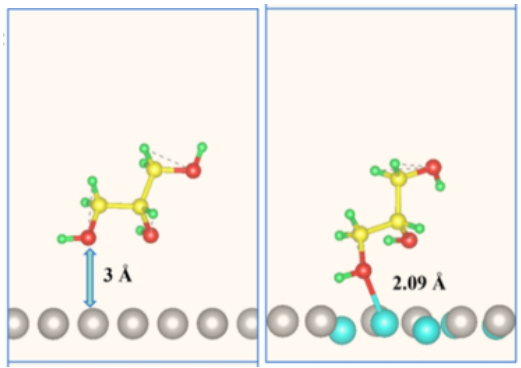

DOI:10.1021/acscatal.4c00483
吸附能计算
吸附能负值越小,吸附越牢固(单位:eV)。柱状图对比不同DFT方法计算的吸附能。不同计算方法对比含范德华力修正的方法(如vdW-DF2)最接近实验值,误差;而传统PBE(灰色柱)低估吸附能,因忽略分子间弱作用力。对弱吸附分子(如CH₄),必须选vdW修正方法,否则会误判催化剂活性。
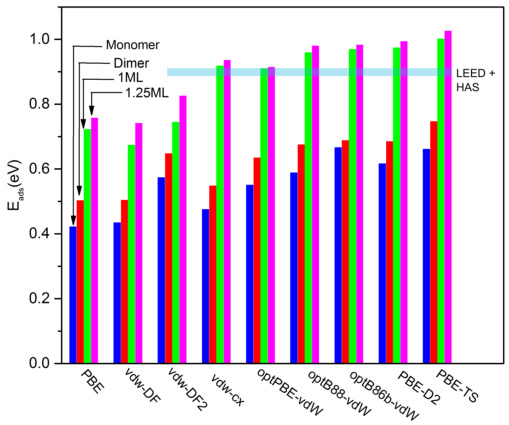
DOI: 10.1063/1.4971790
电子结构分析
通过电子转移、轨道杂化解释“为什么吸附”。Pd掺杂NiCo₂O₄催化生物质转化。电荷密度差图(图b):黄色区域(电荷积累)集中在Pd原子周围,显示Pd从HMF分子“抢”电子,削弱C-H键。
d带中心理论:Pd的d带中心从-1.5 eV上移到-0.8 eV(更接近费米能级),增强反键轨道填充,使HMF吸附能从-1.2 eV降至-2.1 eV。Pd掺杂(右)提升电子转移效率。
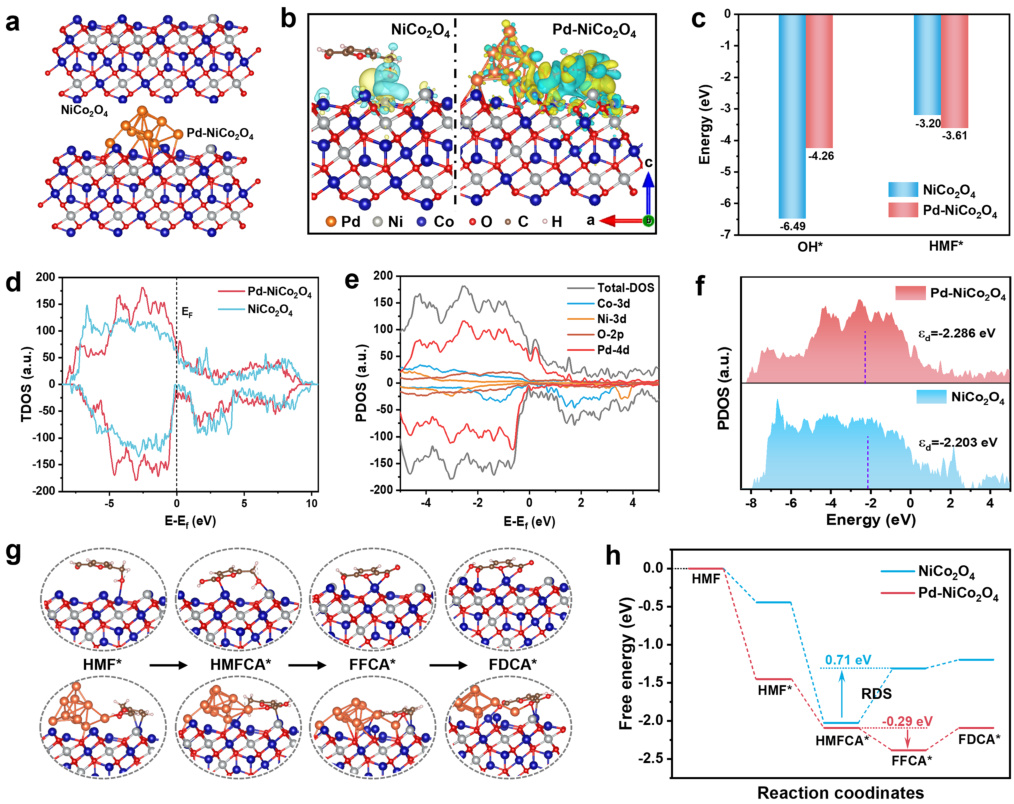
DOI:10.1007/s40820-024-01493-3
反应路径模拟
预测反应能垒,定位决速步。CoO催化CO₂加氢制甲烷。反应网络图+能垒标注(单位:eV)绿色路径最可行CO₂→COOH→CO→CHO→CH₃→CH₄(*代表吸附态)。
决速步:CO→CHO的能垒高达1.8 eV(需高温克服)。关键发现:CH₃与表面O原子结合需0.3 eV,而直接加氢仅需0.1 eV→设计富氢表面可加速反应。
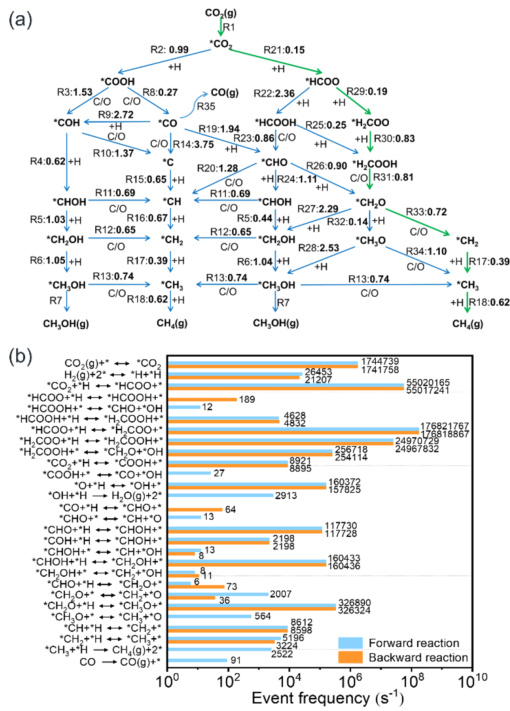
DOI:10.1021/jacsau.2c00632