LAMMPS 是一款功能强大的分子动力学模拟软件,在材料科学、物理学、化学、生物学等多个领域都有广泛的应用。以下是对 LAMMPS 软件的详细介绍:
名称与开发者:LAMMPS 是 Large – scale Atomic/Molecular Massively Parallel Simulator 的简称,即大规模原子分子并行模拟器。
它由美国 Sandia 国家实验室开发,以 GPL license 发布,这意味着源代码开放且免费使用,使用者可根据自身需求对源代码进行修改。
主要特点:LAMMPS 支持并行计算,采用分布式内存 MPI(Message Passing Interface),通过对模拟域进行空间分解实现并行。
它用 C++ 编写,具有良好的可扩展性,可选的库包括 MPI 和单处理器 FFT(Fast Fourier Transform),易于添加新功能和特性。
LAMMPS 通过输入脚本运行,具备定义和使用变量、公式的语法,以及循环和跳出循环的语法,还能从一个脚本同时(并行)运行一个或多个模拟。
材料科学:在金属材料研究中,可模拟金属的力学性能、位错运动、扩散过程等,帮助理解金属在不同条件下的行为,为材料设计和性能优化提供依据。
对于高分子材料,能模拟聚合物链的构象变化、玻璃化转变、结晶过程等,有助于开发新型高分子材料和改进加工工艺。
在纳米材料领域,可研究碳纳米管、石墨烯等纳米材料的力学、电学、热学性质,以及纳米复合材料的界面相互作用和性能。
物理学:用于研究液体的微观结构和动力学性质,如液体的密度分布、扩散系数、黏度等。在固体物理中,可模拟晶体的晶格振动、热导率、相变等现象,深入理解固体材料的物理性质。
还能对软物质体系,如胶体、液晶等进行模拟,研究其自组装行为、流变性质等。
化学:在化学反应动力学研究方面,可模拟化学反应过程中分子的碰撞、反应路径、过渡态等,为化学反应机理的研究提供微观视角。
对于溶液中的化学过程,如电解质溶液的离子分布、化学反应在溶液中的速率等,LAMMPS 也能进行有效的模拟。同时,在表面化学领域,可研究分子在固体表面的吸附、催化反应等过程。
生物学:能够对蛋白质、DNA、RNA 等生物大分子进行模拟,研究其折叠、构象变化、与配体的相互作用等,有助于理解生物大分子的结构与功能关系。
还可用于模拟生物膜的结构和动力学,如脂质分子的排列、膜蛋白的嵌入等,为生物膜相关的研究提供支持。此外,在分子水平上模拟细胞的力学性质、细胞内的物质运输等过程也有应用。
1. 获取 LAMMPS:LAMMPS 的源代码可从其官方网站或 GitHub 仓库获取。从官方网站下载时,访问LAMMPS 官网,导航到下载页面,选择适合操作系统的版本进行下载。下载完成后,解压文件到选择的目录。
2. 安装依赖库:LAMMPS 可能依赖一些其他库,如 MPI 库(用于并行计算)和 FFT 库(用于某些算法)等。如果系统中没有安装这些库,需要先进行安装。
以 Linux 系统为例,安装 MPICH(一种常用的 MPI 实现),可通过以下步骤:
首先下载 MPICH 的源代码包,解压后进入解压目录,执行./configure –prefix=/opt/mpich – gnu进行配置,指定安装路径,然后执行make进行编译,最后执行make install完成安装。
安装 FFTW(一种快速傅里叶变换库)也类似,先下载源代码包,解压后进入目录,执行./configure –prefix=/opt/mathlib/fftw215 – gnu –enable – float等配置选项,再执行make和make install。
3. 编译 LAMMPS:进入解压后的 LAMMPS 源代码目录,通常有多种编译方式可供选择。
可以使用传统的 Makefile 方式,根据自己的需求编辑 Makefile 文件,指定编译器、编译选项以及依赖库的路径等信息,然后执行make命令进行编译。
也可以使用 CMake 工具进行编译,先安装 CMake,然后在 LAMMPS 源代码目录下创建一个构建目录,进入构建目录,执行cmake..命令进行配置,生成 Makefile 文件,再执行make进行编译。
编译完成后,会生成可执行文件lmp,这就是 LAMMPS 的主程序。
分子动力学基本原理:分子动力学是基于牛顿力学,通过求解多体系统中粒子的运动方程来模拟分子的运动和相互作用。
在 LAMMPS 中,每个粒子被视为一个具有质量、位置和速度的实体,根据牛顿第二定律(F = ma),计算粒子受到的力,进而得到粒子的加速度,通过数值积分方法更新粒子的位置和速度,从而模拟粒子在相空间中的运动轨迹。
力场与相互作用势能:为了计算粒子间的相互作用力,LAMMPS 提供了多种力场和相互作用势能函数。
常见的有 Lennard – Jones 势能,用于描述中性原子或分子间的范德华力和短程排斥力;Morse 势能,能更好地描述双原子分子的振动特性;还有用于描述化学键的谐振子势能,以及用于描述静电相互作用的 Coulomb 势能等。
通过选择合适的力场和势能函数,LAMMPS 可以准确地模拟不同类型体系中粒子间的相互作用。
系综与统计力学:LAMMPS 支持多种系综,如微正则系综(NVE,粒子数、体积和能量守恒)、正则系综(NVT,粒子数、体积和温度恒定)、等温等压系综(NPT,粒子数、压力和温度恒定)等。
在不同的系综下,系统的宏观性质和统计分布不同。通过在特定系综下进行模拟,可以计算系统的各种热力学性质,如内能、熵、自由能等,这些性质可以通过对模拟轨迹进行统计平均得到,从而建立起微观分子运动与宏观热力学性质之间的联系。
数值积分算法:在求解粒子运动方程时,需要使用数值积分算法来更新粒子的位置和速度。LAMMPS 中常用的是 Velocity – Verlet 积分算法,它具有较好的数值稳定性和精度,能够准确地模拟粒子的运动。
该算法将粒子的位置和速度更新分为两步,先根据当前的加速度更新速度,再根据更新后的速度更新位置,通过迭代计算得到粒子在不同时刻的状态。
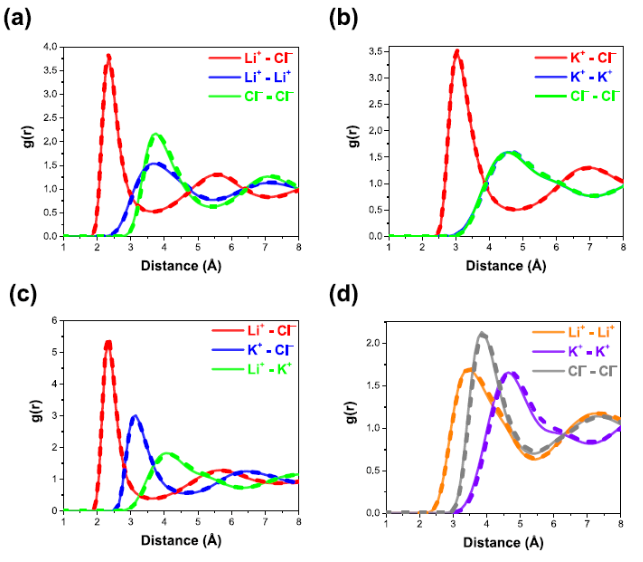
平衡结构确定:LAMMPS 能模拟体系随时间的演化,使原子或分子达到平衡状态,从而确定体系的平衡结构。
例如在模拟金属合金时,通过长时间的模拟,原子会自发调整位置,最终形成能量最低的稳定结构,这有助于研究合金的晶体结构和相组成。
晶体结构性质:对于晶体材料,可计算晶格常数、原子坐标等结构参数。在研究新型半导体晶体时,准确计算晶格常数对于了解其电子结构和光学性质至关重要。
此外,还能模拟晶体的缺陷结构,如空位、位错等,研究这些缺陷对材料性能的影响。
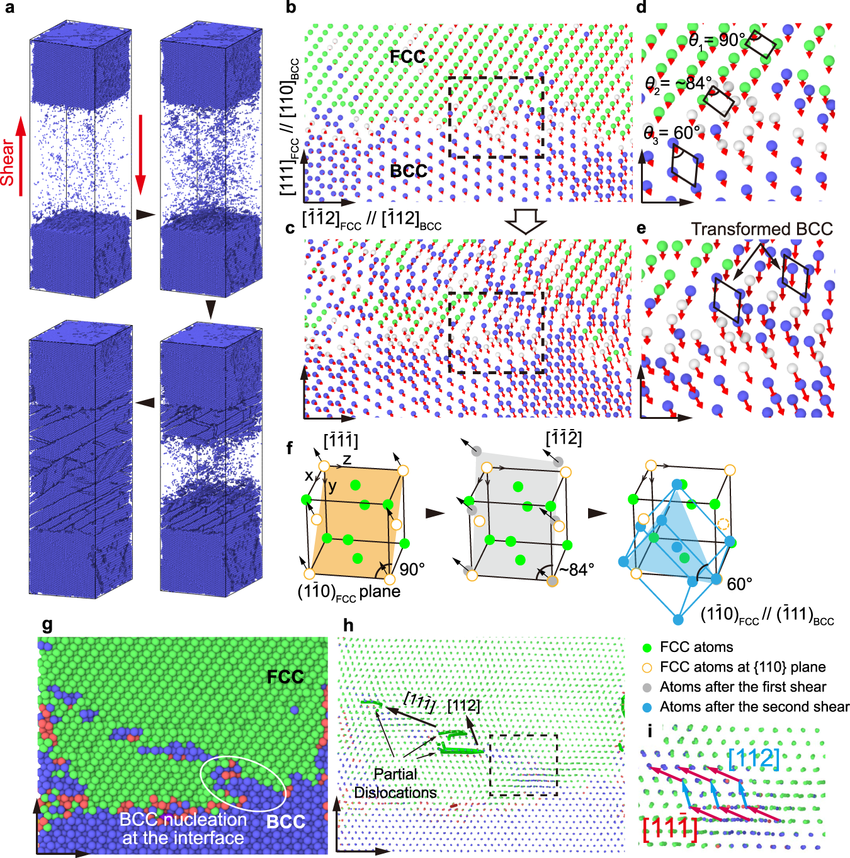
分子动力学模拟:这是 LAMMPS 的核心功能之一。通过牛顿运动定律,模拟原子或分子在一段时间内的运动轨迹。
在模拟生物分子(如蛋白质)时,可观察到蛋白质分子的构象变化,研究其折叠、解折叠过程以及与配体的相互作用机制。
扩散系数计算:根据原子或分子的运动轨迹,LAMMPS 可以计算它们的扩散系数。在研究液体或固体中的扩散现象时,扩散系数是一个重要的参数。例如在锂离子电池电极材料中,锂离子的扩散系数直接影响电池的充放电性能。
黏度计算:对于流体体系,可模拟流体分子的运动,计算流体的黏度。在研究高分子溶液或熔融聚合物的加工性能时,黏度是一个关键参数,LAMMPS 的模拟结果有助于优化加工工艺。
能量计算:能够计算体系的总能量,包括动能、势能等。势能又可细分为不同类型的相互作用能,如范德华能、静电能等。
在研究化学反应时,通过比较反应物和产物的能量,可判断反应的热效应,预测反应的可行性。
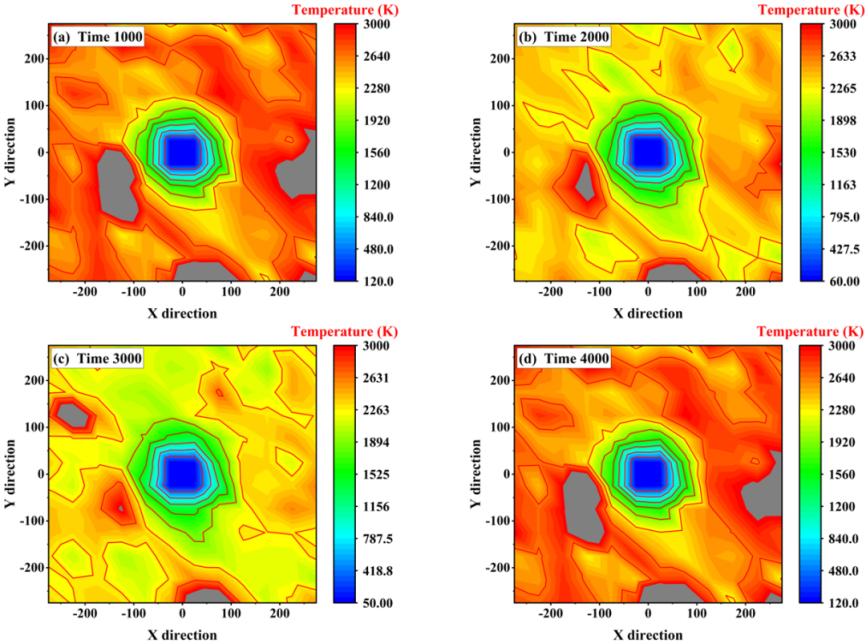
温度和压力计算:模拟过程中可以实时监测体系的温度和压力。在模拟高压下材料的相变过程时,准确控制和计算压力对于理解相变机制非常重要。
同时,温度的变化也会影响分子的运动和相互作用,进而影响体系的结构和性质。
热容计算:通过对体系能量随温度变化的模拟,LAMMPS 可以计算体系的热容。热容是物质的重要热力学性质之一,对于研究材料的热稳定性和热传导性能具有重要意义。
弹性常数计算:对于固体材料,可模拟材料在不同方向上的受力和变形情况,计算其弹性常数。
弹性常数反映了材料抵抗弹性变形的能力,是设计和评估材料力学性能的重要参数。例如在航空航天领域,对材料弹性常数的精确计算有助于优化飞行器结构设计。
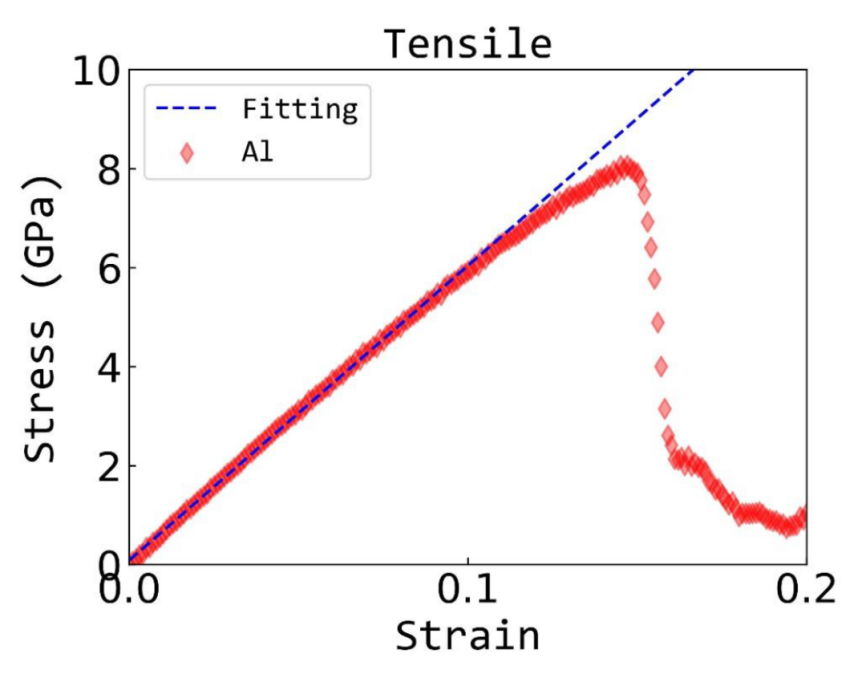
应力 – 应变关系:通过对材料施加外部应力,模拟材料的变形过程,得到应力 – 应变曲线。
这对于研究材料的力学行为,如塑性变形、断裂等现象非常重要。在金属材料加工过程中,了解材料的应力 – 应变关系可以优化加工工艺,提高材料的性能。
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!