

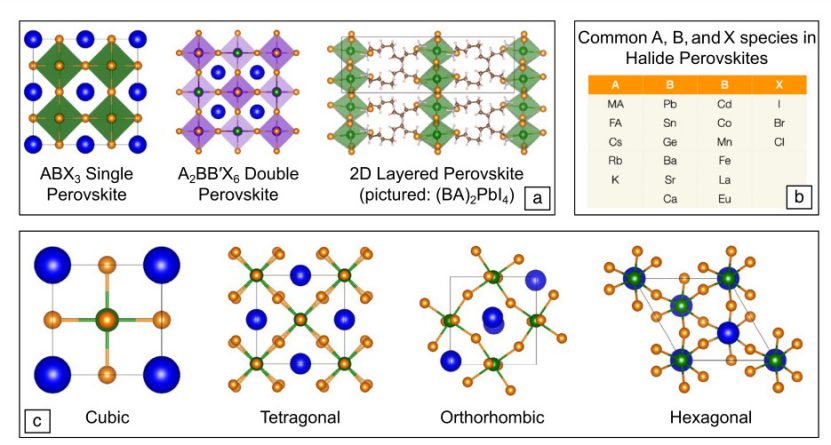
钙钛矿材料通常为ABX₃型结构,其中:
A位:有机阳离子(如甲胺MA⁺、甲脒FA⁺)或无机阳离子(如Cs⁺、Rb⁺);
B位:过渡金属(如Pb²⁺、Sn²⁺);
X位:卤素阴离子(如I⁻、Br⁻、Cl⁻)。
典型材料:
MAPbI₃(甲胺铅碘钙钛矿):广泛应用于光伏领域,具有高光吸收系数。
FAPbI₃(甲脒铅碘钙钛矿):比MAPbI₃更宽的光吸收范围。

Yang J, Mannodi-Kanakkithodi A. High-throughput computations and machine learning for halide perovskite discovery[J]. MRS Bulletin, 2022, 47(9): 940-948.
甲基铅溴钙钛矿(CH₃NH₃PbBr₃ ):属于有机 – 无机混合卤化物钙钛矿,具有良好的光学和电学性能。
在室温下,它具有较高的载流子迁移率和较长的载流子扩散长度,对蓝光具有较强的吸收能力,使其在蓝光发光二极管和蓝光光电探测器等光电器件中具有潜在的应用价值。
同时,它也是研究钙钛矿材料结构与性能关系的重要模型体系之一,通过对其晶体结构、电子结构和光学性质的研究,可以深入理解钙钛矿材料的内在物理机制,为开发新型钙钛矿光电器件提供理论基础。
钙钛矿硫化物(如 CsPbS₃ ):是全无机钙钛矿材料的一种,具有独特的晶体结构和物理性质。与卤化物钙钛矿相比,硫化物钙钛矿通常具有更好的化学稳定性和热稳定性,在高温和恶劣环境下能保持相对稳定的性能。
在光电领域,CsPbS₃表现出良好的光电性能,如较高的载流子迁移率和对近红外光的吸收特性,使其在近红外光电探测器、发光器件以及太阳能电池等方面具有潜在的应用前景。
此外,由于其组成元素相对丰富且环境友好,CsPbS₃钙钛矿材料在大规模应用方面具有一定的优势,受到了越来越多的关注。
钛酸钙(CaTiO₃ ):是典型的氧化物钙钛矿材料,具有立方晶系结构(在高温下),在低温时会发生结构相变,转变为正交晶系或其他晶系结构。
CaTiO₃具有优异的介电性能,其介电常数较高,且在一定温度范围内保持相对稳定,因此在电子陶瓷领域被广泛应用于制作多层陶瓷电容器、微波介质材料等。
此外,CaTiO₃还具有良好的铁电性能和压电性能,在铁电存储器、压电传感器等器件中也展现出潜在的应用价值。
同时,作为一种重要的功能材料,CaTiO₃的晶体结构和物理性质研究为深入理解氧化物钙钛矿材料的结构 – 性能关系提供了基础,对开发新型氧化物钙钛矿功能材料具有重要的指导意义。

如何增加理论计算?
使用VASP软件进行几何优化和电子结构计算,预测焓变、带隙、电荷密度等。
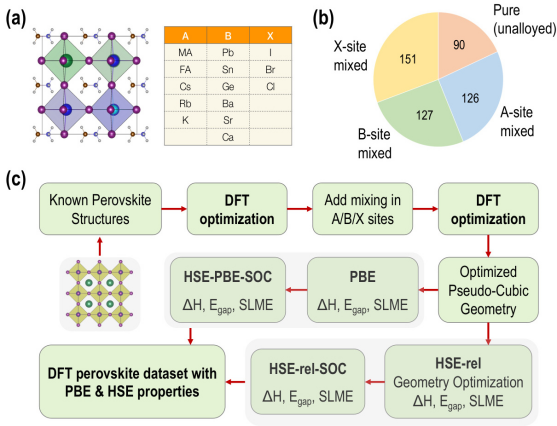
缺陷形成能计算:在钙钛矿材料中,本征缺陷的存在会对其电学、光学和稳定性等性能产生重要影响。计算本征缺陷形成能是理解缺陷行为和材料性能关系的关键步骤。本征缺陷主要包括空位、间隙原子、反位原子等。
例如,在甲基铵铅碘钙钛矿(CH₃NH₃PbI₃ )中,可能存在碘空位(VI )、铅空位(VPb )、甲基铵空位(VMA )等空位缺陷,以及碘间隙原子(Ii )、铅间隙原子(Pbi )等间隙缺陷,还有甲基铵与铅的反位缺陷(MAPb )等。
计算这些本征缺陷形成能可以帮助我们了解缺陷形成的难易程度。较低的缺陷形成能意味着该缺陷在材料中更容易形成。
近年来,研究人员关注添加剂对钙钛矿缺陷形成能的影响。添加剂可以与钙钛矿材料发生相互作用,改变其电子结构和晶体结构,从而影响缺陷形成能。
一些有机小分子添加剂能够与钙钛矿表面的缺陷位点结合,增加缺陷形成能,使缺陷难以形成,起到钝化缺陷的作用,减少非辐射复合,提高材料的光电性能。
理论计算可以详细分析添加剂与钙钛矿之间的相互作用机制,预测不同添加剂对缺陷形成能的影响,为实验中选择合适的添加剂提供理论指导。
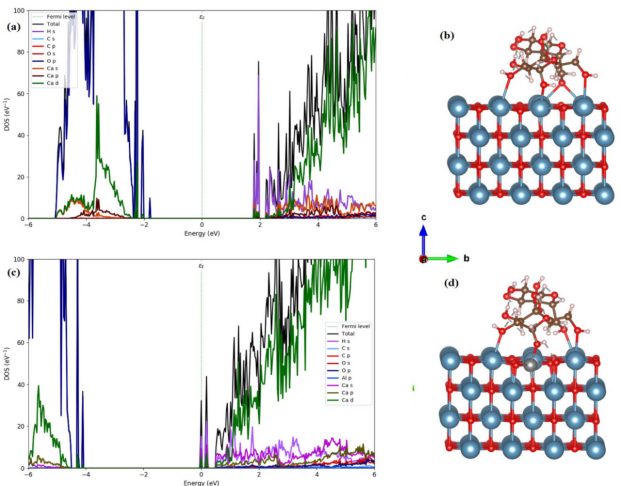
电子结构分析:电子结构决定了钙钛矿中电子的分布和能量状态,进而影响材料的导电性、光学吸收、发光等性能。
通过理论计算得到的钙钛矿能带结构,我们可以了解导带和价带的位置、带隙大小以及能带的色散关系。
直接带隙的钙钛矿材料在光吸收和发射过程中具有较高的效率,因为电子在导带和价带之间跃迁时不需要声子的参与,能量损失较小,这使得它们在太阳能电池和发光二极管等光电器件中具有潜在的应用优势。
而间接带隙的钙钛矿材料,由于电子跃迁需要声子辅助,光吸收和发射效率相对较低。带隙大小决定了钙钛矿对光的吸收范围和能量转换效率,通过调整钙钛矿的组成和结构,可以实现对带隙的调控,以满足不同光电器件的需求。
例如,在钙钛矿太阳能电池中,选择合适带隙的钙钛矿材料可以使其更好地吸收太阳光,提高光电转换效率。
态密度(DOS)分析:是研究电子结构的另一种重要方法。态密度表示在单位能量间隔内电子态的数目,通过计算态密度,可以了解电子在不同能量状态下的分布情况。
在费米能级附近的态密度对材料的电学性质影响较大,较高的态密度意味着更多的电子参与导电,材料的导电性较好。
在光学性质方面,态密度与光吸收和发射过程密切相关,不同能量状态下的电子跃迁对应着不同波长的光吸收和发射,通过分析态密度可以解释钙钛矿的光学光谱特征。
能带结构与态密度(DOS):能带图显示价带顶(VBM)和导带底(CBM),带隙决定光吸收范围。分波态密度(PDOS)揭示各原子轨道对能级的贡献。
DOI: 10.1039/d2ra06340a

吸附能计算及分析
吸附能计算:通过DFT计算,AEP分子在SnO₂表面的吸附能为 -4.75 eV,当SnO₂表面存在氧空位时,吸附能进一步增强,表明AEP分子对缺陷位点具有更强的亲和力。
AEP的膦酸基团(-PO₃H₂)与SnO₂表面的羟基(-OH)发生反应,形成稳定的化学键,抑制表面氧空位和载流子陷阱。
AEP与钙钛矿表面的相互作用,氨基(-NH₂)的吸附:氨基与钙钛矿表面(Pb-I界面)的吸附能为 -0.70 eV,表明较弱的物理吸附作用。
当钙钛矿表面存在铅空位(Vₚ₆)或FA⁺/I⁻反位缺陷时,吸附能显著增加,说明AEP分子可优先吸附于缺陷位点,抑制缺陷形成。
https://doi.org/10.1002/adma.202406532
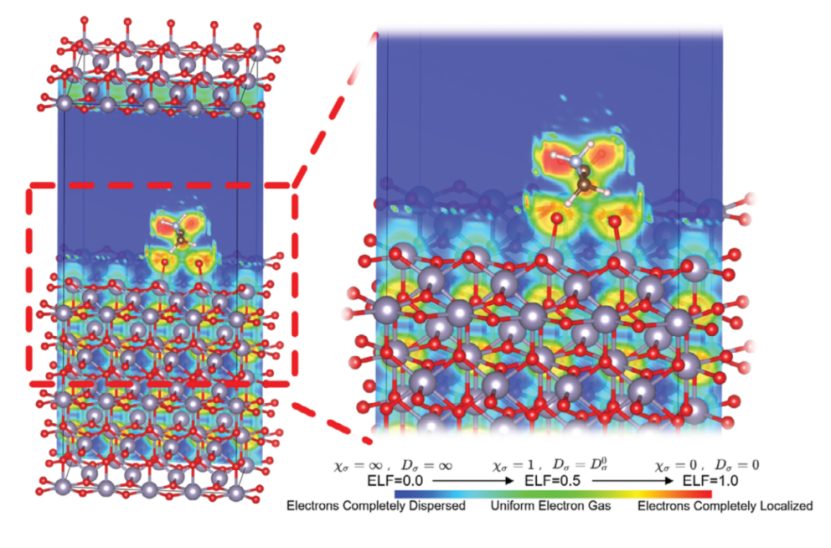
差分电荷密度分析
AEP/SnO₂体系:差分电荷密度图显示,电子从AEP的膦酸基团向SnO₂表面转移,形成电子密度增加的黄色区域(Sn-O键区域),而AEP分子周围电子密度减少(青色区域)。
纵向积分曲线:进一步显示电荷转移主要集中在Sn-O界面(距离SnO₂表面约2 Å范围内),验证了化学键的形成。
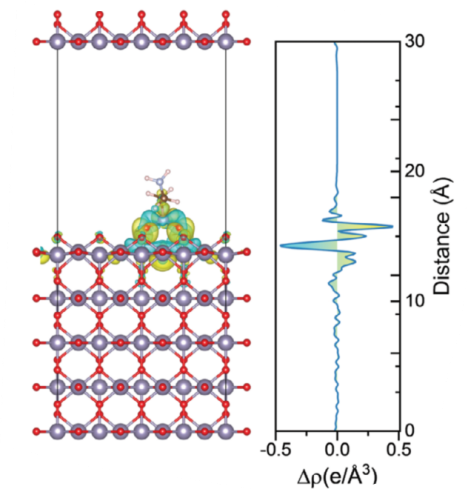
AEP/钙钛矿体系,氨基与钙钛矿中氨基的N原子与钙钛矿表面的I⁻离子形成静电相互作用,电子从氨基向I⁻转移(黄色区域),局部电荷重新分布。AEP分子通过膦酸基团与SnO₂形成化学键,通过氨基与钙钛矿缺陷位点形成静电作用,双重调控界面电荷分布。
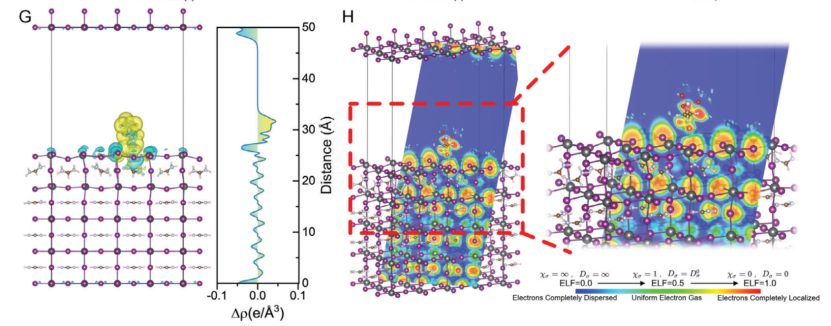
文献通过DFT计算系统揭示了AEP分子在SnO₂/钙钛矿界面的双重作用:
强化学吸附:膦酸基团锚定SnO₂表面,抑制氧空位和漏电流。
静电调控:氨基修饰钙钛矿缺陷位点,降低界面缺陷密度。
差分电荷和ELF分析从电子尺度阐明了电荷转移与局域化机制,为高性能钙钛矿太阳能电池的设计提供了理论指导。
未来需结合多尺度模拟和原位实验,进一步优化界面工程策略。