



Part.01
研究亮点
碱性条件下析氢反应(HER)的催化机理至今仍缺乏原子尺度的明确认识,阻碍了高效催化剂的理性设计。中国科学技术大学国家同步辐射实验室韦世强教授、姚涛教授团队聚焦于该关键问题,构建了结构均一的钴单原子位点(Co₁–N₄)电催化剂,结合Operando XAFS(原位X射线吸收精细结构)技术,首次原子级揭示了HER过程中催化位点的电子结构与配位环境演化。研究发现,在实际工作条件下,Co₁–N₄位点与OH⁻发生作用,形成高价态的HO–Co₁–N₂活性构型,并进一步促进水分子的吸附与活化。理论计算表明,该构型能显著降低Volmer步骤的能垒,从而增强氢析出性能。该工作不仅识别了真实反应条件下的活性中心结构,还提出了通过配位工程调控单原子催化位点的策略,为设计高性能单原子催化剂提供了重要思路,对碱性水电解与清洁能源催化领域具有深远影响。
Part.02
图文解析
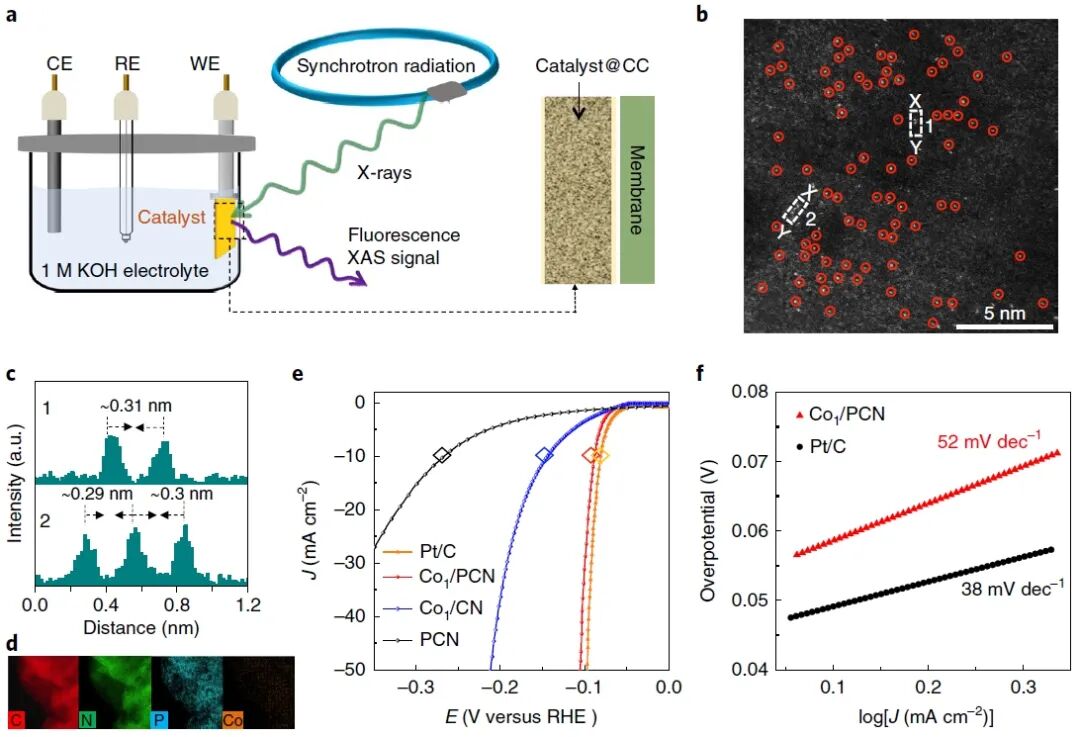
图1:钴单原子电催化剂Co₁/PCN的原位电化学测量装置、微观结构及HER性能
图1展示了用于HER研究的Co₁/PCN催化剂的微观结构表征、电化学性能以及原位表征装置。图1a展示了原位X射线吸收精细结构(XAFS)测量的电化学装置示意图,其中包括工作电极(WE)、对电极(CE)、参比电极(RE)等电极构成,以及X射线照射和荧光信号收集路径。这一装置实现了在实际工作电位下的同步XAFS测量,确保可探测活性位点的结构演变。图1b为HAADF-STEM图像,显示了钴以单原子形式(红圈标记)均匀分布在PCN载体上,且未观察到团簇或纳米颗粒,验证了催化剂的单原子结构。图1c展示了图1b图像中X–Y方向的强度剖面图,Co原子间距约为0.29 nm,远大于Co原子直径,进一步证明其为分散单原子状态。图1d为元素分布图,展示了C、N、P和Co元素在PCN载体中的均匀分布,且磷元素主要来源于PCN的磷化改性。图1e展示了在1.0 M KOH电解液中不同催化剂的HER极化曲线。Co₁/PCN表现出显著优于空白PCN和Co₁/CN样品的催化活性,其@10 mA cm⁻²时过电位仅为89 mV,接近商业Pt/C性能。1f为Tafel曲线,显示Co₁/PCN的Tafel斜率为52 mV dec⁻¹,表明其遵循Volmer–Heyrovsky机制,反应动力学优异。综合来看,图1确立了Co₁/PCN的单原子结构和优异的HER性能,为后续原位结构研究奠定基础。
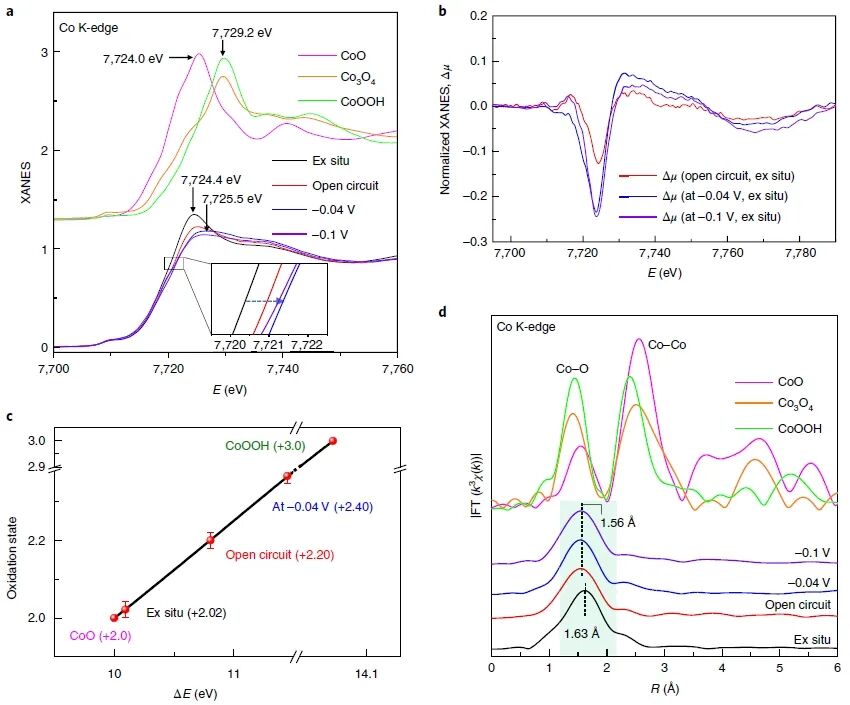
图2:原位XAS测量揭示Co₁/PCN催化剂在HER过程中的结构与氧化态演化
图2展示了通过原位X射线吸收谱(XAS)技术研究Co₁/PCN催化剂在碱性HER条件下电子结构和局部配位环境的动态变化。图2a展示了不同工作电位下Co K-edge XANES(X射线近边吸收结构)光谱的变化。在开路条件下,XANES谱线的吸收边相比ex situ样品有明显向高能方向移动,表明Co原子的氧化态升高;而在工作电位(−0.04 V与−0.1 V)下,这一吸收边进一步移动,暗示催化过程中Co原子经历了更高程度的氧化。对比标准样品CoO、Co₃O₄与CoOOH的谱线位置,推测Co从+2价逐步上升至+2.4左右。图2b为不同状态下的XANES差谱图,更直观地揭示了吸收边位移与谱形变化,进一步确认了氧化态提升与Co配位环境的变化。图2c定量给出了不同状态下Co的平均氧化态:ex situ为+2.02,开路条件为+2.20,−0.04 V下为+2.40。这一结果表明,Co单原子位点在碱性电解质中会受到OH⁻吸附诱导产生高价态结构,成为HER活性中心。图2d展示了不同条件下的Co K-edge EXAFS(扩展X射线吸收精细结构)傅里叶变换谱。与ex situ状态下主要的Co–N配位峰相比,开路与工作条件下主峰向低R值位移(由1.63 Å变为1.56 Å),且强度发生变化,说明配位环境发生重构,出现了Co–O配位,印证了OH⁻或H₂O在Co位点的吸附现象。
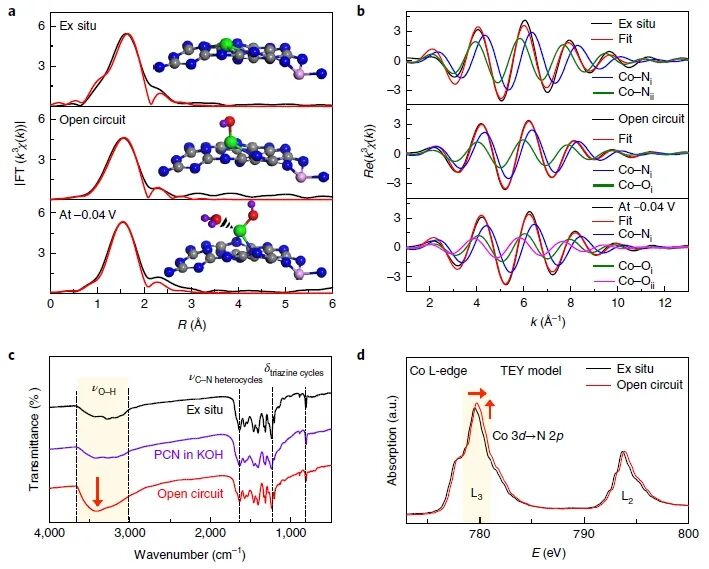
图3:结构分析揭示Co单原子位点在HER过程中的配位环境与电子态重构
图3展示了通过EXAFS拟合、FTIR和软X射线吸收等多种手段,对Co₁/PCN在HER过程中的原子级配位结构和电子态演化进行深入解析。图3a显示了不同条件下的Co K-edge EXAFS傅里叶变换谱的一壳层拟合结果。对ex situ样品拟合表明,Co以四配位形式结合N原子,形成典型的Co₁–N₄结构;在开路条件下,拟合结果变为Co₁–N₂–O₁结构,即两个Co–N配位(R ≈ 2.01 Å)和一个Co–O配位(R ≈ 2.08 Å),说明OH⁻开始吸附;而在工作电位(−0.04 V)下,则进一步演化为Co₁–N₂–O₂结构,即两个N配位和两个O配位,其中一个O原子归因于吸附的水分子,反映出典型的**HO–Co₁–N₂与H₂O–(HO–Co₁–N₂)**结构形成。这种结构变化清晰反映出催化过程中的位点演化,验证了HER活性来自该动态生成的高价态构型。图3b为对应的k³χ(k)振荡拟合图,红色拟合曲线与黑色实验曲线高度吻合,进一步确认所提拟合模型的准确性与合理性。图3c为不同样品在开路条件下的FTIR谱图。相比ex situ样品和空白PCN,Co₁/PCN样品在3000–3500 cm⁻¹范围内出现显著增强的–OH伸缩振动峰,表明有大量OH⁻物种吸附于Co位点。结合XANES结果,这些–OH峰更可能归因于化学吸附的HO–Co结构,而非H₂O物理吸附。图3d进一步通过Co L₂,₃边的软X射线吸收谱(TEY模式)验证了电子结构变化。与ex situ样品相比,开路状态下的L₃峰显著增强并发生0.2 eV蓝移,表明Co 3d态空轨道增多,电子密度下降,进一步印证Co氧化态的提升。
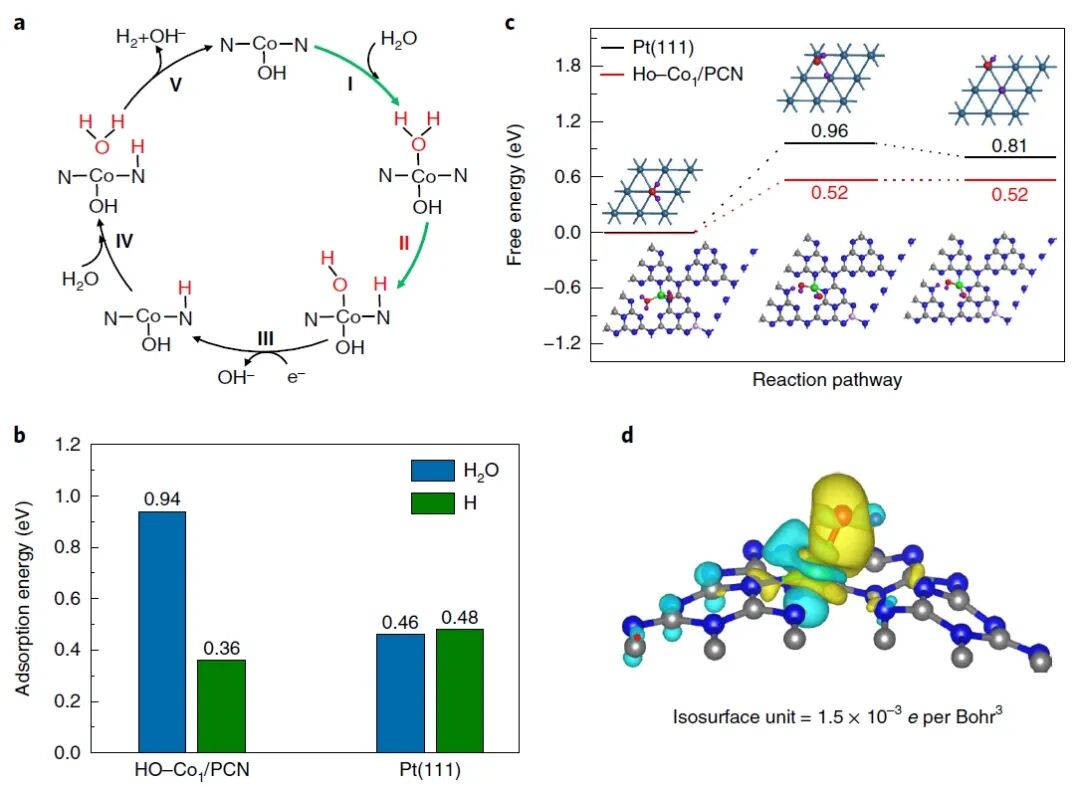
图4:理论研究揭示HO–Co₁/PCN催化剂在碱性HER中的反应机理与水分子解离机制
图4展示了基于密度泛函理论(DFT)计算对Co₁/PCN催化剂在碱性HER中的反应机理进行的理论研究,重点分析了水分子解离(Volmer步骤)和氢吸附的动力学过程。图4a展示了在碱性HER中,HO–Co₁/PCN催化剂的反应循环。首先,水分子(H₂O)吸附到Co位点并解离为OH⁻与H,随后H与另一个水分子中的H*发生反应,最终生成氢气(H₂)。这一过程的第一个关键步骤是水分子在Co位点的吸附和解离,这被认为是碱性HER中的Volmer步骤**,通常是反应的速率决定步骤。图中的步骤I至V显示了这一反应过程的详细步骤,表明水解离反应的速率受限于Co位点的水吸附能力与水解离的能垒。图4b展示了水分子吸附能的计算结果,比较了HO–Co₁/PCN与商业Pt(111)催化剂的水分子吸附能。结果显示,HO–Co₁/PCN表面水分子的吸附能为0.94 eV,高于Pt(111)的0.46 eV,表明HO–Co₁/PCN在水分子吸附方面更具优势,进而促进水的解离过程。图4c展示了水分子解离步骤的能垒计算。图示中,HO–Co₁/PCN的解离能垒为0.52 eV,明显低于Pt(111)上的0.96 eV,表明在HO–Co₁/PCN催化剂上,水分子的解离更加容易且放热,能够提供更快的质子供应,从而加速氢气的生成。这一计算结果与实验发现一致,进一步证实了HO–Co₁/PCN在碱性条件下表现出优异的HER性能。图4d展示了HO–Co₁/PCN的电荷密度差图,显示Co原子通过轨道混合将电子供给给邻近的N原子和OH⁻离子。这一电荷转移过程加强了Co–OH的吸附作用,有助于促进水解离反应的发生。通过Bader电荷计算,计算表明在开路条件下Co的氧化态由+2.05增加到+2.16,进一步验证了OH⁻吸附所引发的Co氧化态变化。
Part.03
结论与展望
本研究通过结合原位XAFS谱学技术与密度泛函理论计算(DFT),成功捕捉了钴单原子催化剂在碱性HER条件下的动态结构演化。实验证实,Co₁–N₄在工作电位下可转变为高价态的HO–Co₁–N₂结构,并吸附水分子形成H₂O–(HO–Co₁–N₂)中间体,有效推动水分子的解离(Volmer步骤),从而显著提升催化效率。理论计算进一步验证了该结构的高水吸附能与低解离能垒,并揭示出该过程中强烈的电荷转移行为是HER活性增强的关键。这些结果证实了原子级配位环境调控在提升碱性HER催化活性中的重要作用,也为其他多电子转移反应中的单原子催化机制研究提供了范式参考。
展望未来,本研究不仅为设计结构可控、反应选择性强的SAS(单原子催化剂)奠定了理论与实验基础,也展示了原位表征技术在揭示电催化反应本质中的巨大潜力。后续研究可进一步探索不同过渡金属SAS在复杂反应环境中的演化行为,拓展该策略在氧还原、CO₂还原和氮还原等反应中的应用。此外,引入人工智能辅助的高通量筛选与建模也有望加速此类催化剂的设计与应用推广。
本文源自微信公众号:超分辨电化学
原文标题:《中科大Nature Catalysis:原位同步辐射吸收谱学技术揭示真实反应活性位点》
原文链接:https://mp.weixin.qq.com/s/7lHVvYHTCliostAtxFk5Fg
本转载仅出于分享优质测试干货,旨在传递更多观点,并不代表赞同其全部观点或证实其内容的真实性。文章中所包含的图片、音频、视频等素材的版权均归原作者所有。如有侵权请告知删除。