



表1: 铜表面计算的INCAR文件
SYSTEM = Rhodium surface calculation
INIWAV = 1 electr: 0-lowe 1-rand
BMIX = 2.0
POTIM = 1.0 time-step for ion-motion
SIGMA = 0.2; ISMEAR = 1 broad. in eV, -4-tet -1-fermi 0-gaus
INCAR 文件
INCAR 文件是 VASP 的核心输入文件。它决定 “做什么和怎么做”,并且可以包含相对较多的参数。大多数参数都有方便的默认值,不了解其含义的用户不应更改任何默认值。在处理 INCAR 文件时要非常小心,它是错误和错误结果的主要来源!
INCAR 文件是一个标签无格式的 ASCII 文件:每行由一个标签(即字符串)、等号”=”和若干数值组成。如果每个参数对之间用分号”; “隔开,则可以在一行中给出多个参数对 ( tag = values )。如果一行以反斜线结束,下一行将被视为续行。注释前通常有符号 “#”,但在大多数情况下,注释可以附加到参数-值对中,而不用 “#”。在这种情况下,注释中应避免使用分号。
下文将介绍 INCAR 文件中给出的参数。
尤其是初始化过程可能会有点复杂,请仔细阅读第 6.2 节;其中给出了一些初始化参数如何交互以及如何一起使用的提示。
6.1 所有参数(至少为大部分参数)
下面是目前支持的所有参数的简要概述。经常使用的参数会被特殊显示。
| NGX, NGY, NGZ | 轨道的FFT网格 (参见 6.3,6.11) | |
| NGXF,NGYF,NGZF | 电荷的FFT网格 (参见6.3,6.11) | |
| NBANDS | 计算中包含的能带数 (参见6.5) | |
| NBLK | 阻止某些BLAS调用(参见6.6) | |
| SYSTEM | 体系名称 | |
| NWRITE | 详细写入标签(写入的数量) | |
| ISTART | 任务开始: 0-new 1-cont 2-samecut | |
| ICHARG | 电荷: 1-file 2-atom 10-const | |
| ISPIN | 自旋极化计算 (2-yes 1-no) | |
| MAGMOM | 初始化磁矩/原子 | |
| INIWAV | 初始化电荷波函数: 0-lowe 1-rand | |
| ENCUT | 截断能(单位eV) | |
| PREC | 过程: medium, high or low | |
| PREC | VASP.4.还包括: normal, accurate | |
| NELM, NELMIN and NELMDL | 电子步的nr. | |
| EDIFF | 电子步的停止标准 | |
| EDIFFG | 离子步的停止标准 | |
| NSW | 离子步步数 | |
| NBLOCK and KBLOCK | inner block; outer block | |
| IBRION | 离子弛豫: 0-MD 1-quasi-New 2-CG | |
| ISIF | 计算应力及需要弛豫的部分 | |
| IWAVPR | 平面波预测: 0-non 1-charg 2-wave 3-comb | |
| ISYM | 对称性: 0-nonsym 1-usesym | |
| SYMPREC | 对称性路径过程 | |
| LCORR | 力的Harris校正 | |
| POTIM | 离子位移的时间步长 (fs) | |
| TEBEG, TEEND | 运行温度 | |
| SMASS | Nose质量参数(am) | |
| NPACO and APACO | 距离及nr. of slots for P.C. | |
| POMASS | 离子质量(am) | |
| ZVAL | 离子价态 | |
| RWIGS | Wigner-Seitz半径 | |
| NELECT | 电子总数 | |
| NUPDOWN | 将自旋磁矩固定为指定值 | |
| EMIN, EMAX | DOSCAR文件的能量范围 | |
| ISMEAR | 部分占据: -5 Blochl -4-tet -1-fermi 0-gaus ¿0 MP¨ | |
| SIGMA | 展宽(eV) -4-tet -1-fermi 0-gaus | |
| ALGO | 算法: Normal (Davidson) — Fast — Very Fast (RMM-DIIS) | |
| IALGO | 算法: use only 8 (CG) or 48 (RMM-DIIS) | |
| LREAL | 实空间的非局域投影 | |
| ROPT | 实空间的非局域投影的网格数 | |
| GGA | xc类型: e.g. PE AM or 91 | |
| VOSKOWN | 使用Vosko, Wilk, Nusair 插值 | |
| DIPOL | 偶极子晶胞中心 | |
| AMIX, BMIX | 混合标签 | |
| EBREAK, DEPER | 特殊控制标签 | |
| TIME | 特殊控制标签 | |
| LWAVE,LCHARG, LVTOT, LVHAR | 生成WAVECAR/CHGCAR/LOCPOT | |
| LELF | 生成ELFCAR | |
| LORBIT | 生成PROOUT | |
| NPAR | 带间并行 | |
| LSCALAPACK | 关闭scaLAPACK | |
| LSCALU | 关闭LU 分解 | |
| LASYNC | 将计算与通信重叠 | |
6.2 INCAR 文件中的常用设置
6.2.1 静态计算
只需删除 WAVECAR 文件并从头开始,INCAR 文件中无需设置任何参数。某些参数的默认值为
| ISTART = | 0 # startjob: no WAVECAR file |
| ICHARG = | 2 # charge: from atoms |
| INIWAV = | 1 # random initialization for wf. |
| NELM = | 40 # maximum of 40 electronic steps |
| NELMIN = | 2 # minimum of two steps |
| NELMDL = | -5 # no update of charge for 3 steps |
| EDIFF = | 10E-4 # accuracy for electronic minimization |
在某些情况下,从旧的 WAVECAR 文件开始计算是有意义的(例如,继续弛豫或继续增加截断能 ENCUT)。在这种情况下,只需保留 WAVECAR 文件并启动 VASP 即可。再次强调,也可使用一个空的 INCAR 文件。现在的默认值是:
| ISTART = | 1 | # continue from WAVECAR file |
| ICHARG = | 0 | # charge from orbitals |
| NELM = | 40 | # maximum of 40 electronic steps |
| NELMIN = | 2 | # minimum of two steps |
| NELMDL = | 0 | # immediately update charge |
如果存在旧的 CHGCAR 文件,可以手动设置 ICHARG=1。与默认设置相比,在电荷变化剧烈的情况下,可以节省几步。若要从以前的计算开始继续弛豫,请将 CONTCAR 文件复制到 POSCAR。
6.2.3 建议的最低设置
尽管可以使用空 INCAR 文件执行前面的计算,但建议始终手动设定几个参数:
ISMEAR = 0 or 1 or -5 # method to determine partial occupancies
所有计算都应包含这四个参数。它们完全控制着计算的技术精度,尤其是基组(ENCUT),以及是否使用实空间投影方法。如果两个计算的前三个参数设置相同,则两个计算的总能量只能进行比较和相减。理想情况下,ISMEAR 参数也应在所有计算中保持一致(但在某些情况下可能很难做到)。
6.2.4 从不合理的初始猜测进行高效弛豫
如果想从一个偏离能量最小值的构型开始进行有效弛豫,请在 INCAR 文件中设置以下值(为了简洁起见,建议的手动设置请参见第 6.2.3 节):
| NELMIN = 5 | # do a minimum of four electronic steps |
| EDIFF = 1E-2 | # low accuracy |
| EDIFFG = -0.3 | # accuracy of ions not too high |
| NSW = 10 | # 10 ionic steps in ions |
| IBRION = 2 | # use CG algorithm |
该方法只在开始的几步精度较低,但由于至少要进行 5 步电子运算,因此计算出的电子基态的精度会逐渐提高。如果您是较高级用户,也可以使用阻尼 MD 算法,它通常比 CG 算法更有效:
POTIM = 0.4 # time step needs to chosen with care
这种情况下,过大的 POTIM 会导致不收敛。
6.2.5 从预收敛的起始猜测进行高效弛豫
在接近局部最小值时,可变尺度(RMM-DIIS 算法)最为有效。INCAR 文件((为了简洁起见,建议的手动设置请参见第 6.2.3 节):
NFREE = 10 # estimated degrees of freedom of the system
现在需要非常精确的作用力(EDIFF 较小)。此外,在每次离子位置更新之间至少要进行 8 个电子步,这样电子基态的计算才会非常精确。NELMIN=8 仅适用于电荷波动严重的体系,这种体系很难进行电子收敛。对于大多数体系,NELMIN=4 和 NELMIN=6 之间的值就足够了。
6.2.6 分子动力学
请参见第 9.7 节。
6.2.7 加快计算速度
在 INCAR 文件中使用以下几行,可提高大型体系的 VASP 效率:
LREAL = A # evaluate projection operators in real space NSIM = 4 # blocked algorithm update, four bands at a time
此外,还可以尝试设置 MAXMIX 标签。
6.3 NGX、NGY、NGZ 和 NGXF、NGYF、NGZF-标签
NGX、NGY、NGZ 控制 FFT 网格中沿三个晶格向量方向的网格点数量。X 对应第一个网格矢量,Y 对应第二个网格矢量,Z 对应第三个网格矢量(X、Y 和 Z 与笛卡尔坐标无关,不要被历史上的命名惯例所误导)。
NGXF、NGYF、NGZF 控制第二精细 FFT 网格的网格点数量。如果使用超软(US)Vanderbilt电势或 PAW 方法,则在该网格上表示局域增量电荷。此外,如果(也只有在)使用 US 赝势,还可以在第二精细的 FFT 网格上计算局域电势(交换相关电势、Hartree势和离子电势)。
注意:如果不使用 US 赝势或 PAW 方法,则无需将 NGXF 设置为大于 NGX 的值。在这种情况下,电荷密度和局域电势都不会在精细网格上设置。唯一的结果就是浪费大量存储空间。在这种情况下,只需将 NGXF、NGYF、NGZF 设置为 1。
在 VASP.4.X 中,所有参数都在运行时确定,或者使用默认值(第 6.11 或 5.23 节),或者从 INCAR 文件中读取 NGX 等(见第 6.3 节)。
6.4 KSPACING 标签和 KGAMMA 标签
KGAMMA = [logical]
默认值:
| KSPACING | = | 0.5 |
| KGAMMA | = | .TRUE. |
如果没有 KPOINTS 文件(见第 5.5 节),标签 KSPACING 将决定 k 点的数量。KSPACING 是 k 点之间允许的最小间距,单位为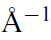 。当间距减小时,k 点的数量会增加。第一、第二和第三倒晶格矢量方向上的 k 点数由以下公式决定:
。当间距减小时,k 点的数量会增加。第一、第二和第三倒晶格矢量方向上的 k 点数由以下公式决定:
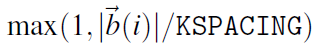
这些值四舍五入到下一个整数。生成的网格要么以 Γ 点为中心(例如包括 Γ 点)(KGAMMA=.TRUE.),要么像Monkhorst Pack网格的做法那样偏离 Γ 点(KGAMMA=.FALSE.)(参阅章节 5.5.3)。默认情况下,网格包括 Γ 点。
6.5 NBANDS标签
NBANDS = [integer]
默认值:
= 0.6*NELECT + NMAG (spin-polarized)
NBANDS 决定计算中的实际能带数。
选择 NBANDS 时,计算中应包含相当数量的空带。至少需要一个空带。否则,VASP 将发出警告。
从技术角度来看,NBANDS 也很重要:在迭代矩阵对角化方案中,接近计算向量数最大值的特征向量收敛速度要比最低特征向量慢得多。如果计算中没有包含足够的空带,可能会导致性能大幅下降。因此我们建议将 NBANDS 设置为 NELECT/2 + NIONS/2,这也是 makeparam 工具和 VASP.4.X 的默认设置。在某些情况下,大型体系也可以在不降低性能的情况下将附加带的数量减少到 NIONS/4,但另一方面,过渡金属需要更多的空带(最多 2*NIONS)。
为了检查这一参数,请在固定电位(ICHARG=12)下进行多次计算,同时增加空带数量(例如,从 NELECT/2 + NIONS/2 开始)。迭代 10-15 次后,精度应达到 10-6。注意 RMM-DIIS 方案(IALGO=48)比默认 CG 算法(IALGO=38)对能带数更为敏感。
6.6 NBLK标签
NBLK = [integer]
默认值:
= 256 in VASP.5.2, if dfast
这决定了许多 BLAS 第 3 级例程的阻塞因子。
在某些情况下,VASP 必须对当前轨道进行单元变换。这需要使用工作数组 CBLOCK 和以下 FORTRAN 代码来完成:
ILEN=MIN(NBLK,NPL-IBLOCK)
200 CONTINUE
100 CONTINUE
ZGEMM 是 BLAS 软件包中的矩阵 × 矩阵乘法命令。该调用执行的任务由写在 ZGEMM 调用上方的注释行表示。一般来说,NBLK=16 或 32 对于超标量机器来说已经足够大了。在矢量机器上,为了获得最佳性能,可能需要一个较大的值(NBLK=128)。
6.7 SYSTEM标签
SYSTEM = [string]
默认值:
SYSTEM = SYSTEM=unknown system.
SYSTEM 标签后是一个可能包含空格的字符串。’title’字符串仅供用户使用,为帮助用户确定要对该特定输入文件做什么。请尽可能详细。字符串被读入并写入主输出文件 OUTCAR。
6.8 NWRITE标签
NWRITE= 0 | 1 | 2 | 3 | 4
默认值: NWRITE=2
该标签决定向文件 OUTCAR 写入多少内容(”verbosity flag”)。
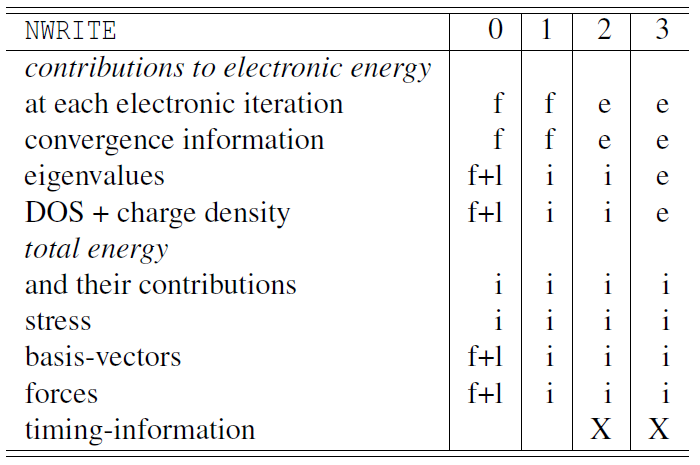
X 可应用时
对于长时间的分子动力学运行,使用 NWRITE=0 或 NWRITE=1。对于短时间运行,使用 NWRITE=2。如果出现问题,NWRITE=3 可能会提供信息。NWRITE=4 仅用于调试。
6.9 ENCUT标签
ENCUT= [real]
默认值
ENCUT = largest ENMAX from POTCAR-file
平面波基组的截断能,单位为 eV。所有动能小于 Ecut 的平面波都包含在基组中即:
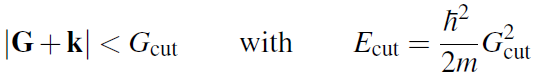
每个 k 点的平面波数量都不相同,因此在能量-体积计算等方面具有更优越的性能。如果体积增大,平面波的总数会发生相对平滑的变化。标准 |G| cut(即每个 k 点的基组相同)会导致非常粗糙的能量-体积曲线,一般来说,这会使能量收敛速度较慢。
从 VASP 3.2 版开始,POTCAR 文件包含默认的 ENMAX(和 ENMIN)行,因此原则上无需在 INCAR 文件中指定 ENCUT。对于有多个种类的计算,将使用最大截断值(ENMAX 或 ENMIN)进行计算(见下文第 6.11 节)。出于一致性考虑,我们仍然建议在 INCAR 文件中手动指定截断值,并在整组计算中保持不变。
6.10 ENAUG标签
ENAUG= [real]
默认值:
ENAUG = EAUG from POTCAR file
增量电荷的动能截断,该行确定了NGXF、NGYF和NGZF(另请参见6.11节)。