

说明:本文系统介绍了电催化剂设计中的关键参数——描述符,涵盖热力学、电子结构、几何结构及材料本征性质四大类描述符。阅读本文,读者能深入了解描述符在理性设计催化剂中的重要性,学会如何结合多参数解析复杂催化体系。
1、热力学描述符(吸附能相关)
中间体吸附自由能 (ΔGads)是最核心、应用最广泛的描述符类别,其中反应决速步中间体的吸附能通常是关键描述符。基于Sabatier原理(最佳催化剂与反应中间体的结合强度应“适中”),通常表现为“火山型”关系图。
氢吸附自由能:ΔGH*是析氢反应(HER)中最常见的描述符。对于Pt(111),ΔGH*≈0,接近火山图顶点。ΔGH*越接近0 eV,HER活性通常越高。
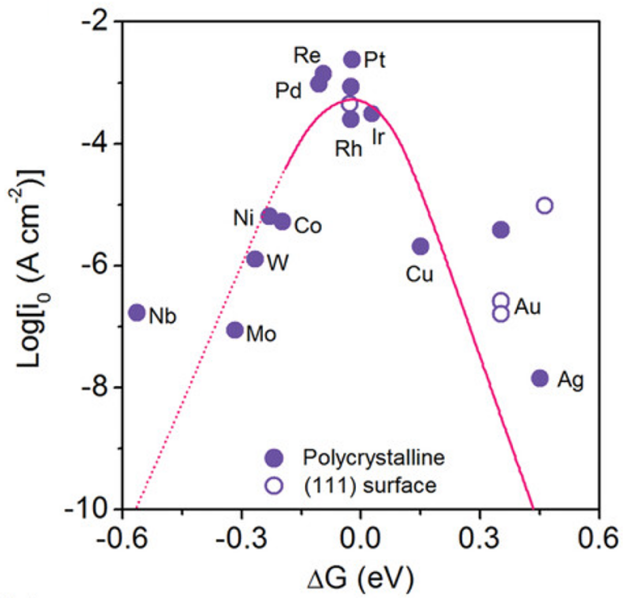

图1 H*的吉布斯自由能和交换电流密度的关系示意图(圆圈代表实验数据,曲线是基于DFT计算的微动力模型的预测,其中的虚线表示金属和H超过0.2 eV/H通常会形成氧化物)。DOI:10.1002/chin.200524023
含氧中间体吸附自由能:OER中常用的吸附自由能有ΔGO,ΔGOH,ΔGOOH*。通常认为ΔGO*–ΔGOH*或理论过电位η=max{ΔGOH,ΔGOOH–ΔGOH*,ΔGO2* – ΔGOOH*}/e–1.23V(忽略动力学和熵)能较好地关联OER活性。理想的催化剂应使这三个步骤的能垒尽可能均衡。ΔGOOH*或ΔGO*常被用作ORR活性的关键描述符(ORR是OER的逆过程,但路径可能不同)。
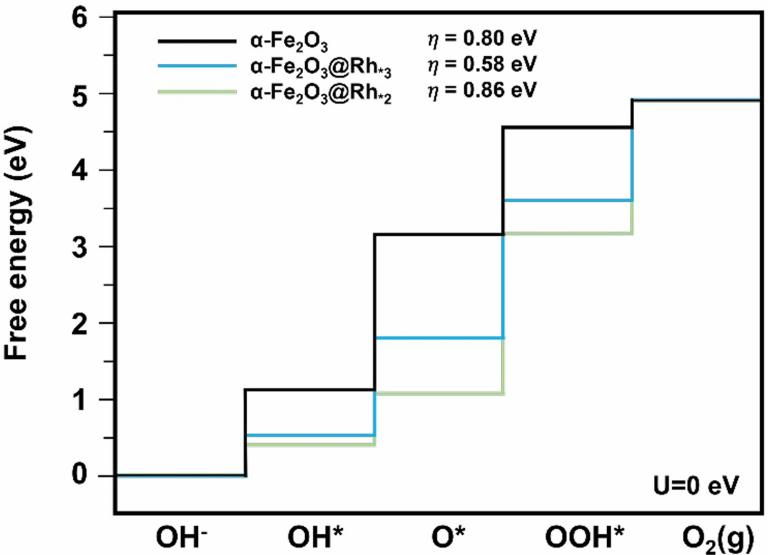
图2 α-Fe2O3@Rh*3和α-Fe2O3@Rh*2的自由能和计算过电位。DOI:10.1021/acsami.3c04458
如果一个反应能改变,其他反应能也会改变。由于中间体之间的标度关系,在金属和金属氧化物表面观察到这种相关能量现象。例如HO*和O*的结合能之间的线性关系,从图3可以看出,HO*和HOO*之间的自由能差几乎是恒定的,与表面结合强度无关。
M. Koper在一篇综述(DOI:10.1016/0254-0584(86)90045-3)中指出,HO*和HOO*的结合在金属和氧化物表面上都以一个约为3.2 eV的常数相互关联,而不考虑结合位置,这意味着HO*和HOO*之间存在普遍的标度关系。此外,不只在OER中,在合成氨反应或氮还原反应中,含氮中间体(如N₂Hₓ*、NHₓ*等)的吸附能同样呈正相关关系。
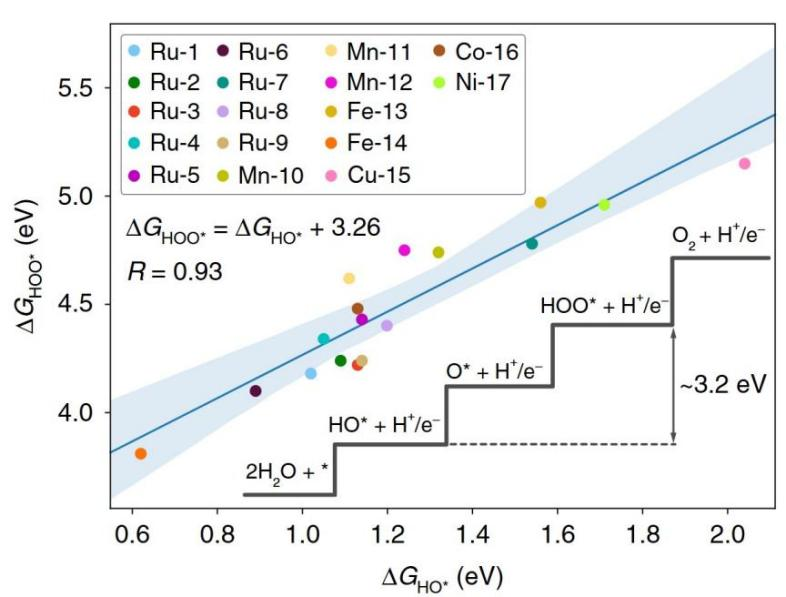
图3 OER催化剂和HO*和HOO*中间体之间的线性标度关系(阴影蓝色区域表示线性模型的99%置信区间)DOI:10.1038/s41467-019-12994-w
氮还原中间体吸附自由能:ΔGN*,ΔGNNH*,ΔGNH2*等。
其他反应中间体吸附自由能:如CO₂还原中的ΔGCOOH*,ΔGCO*,ΔGCHO*等。
2、电子结构描述符
d带中心(εd):对于过渡金属及其化合物极其重要。εd上移(更接近费米能级),d带变窄,与吸附质轨道的反键态填充减少,导致吸附增强;εd下移(远离费米能级):d带变宽,反键态填充增多,导致吸附减弱。
其与ΔGads有很强的关联性,是理解吸附能趋势的重要理论基础。但它是简化模型,对复杂体系(如氧化物)的适用性有限。
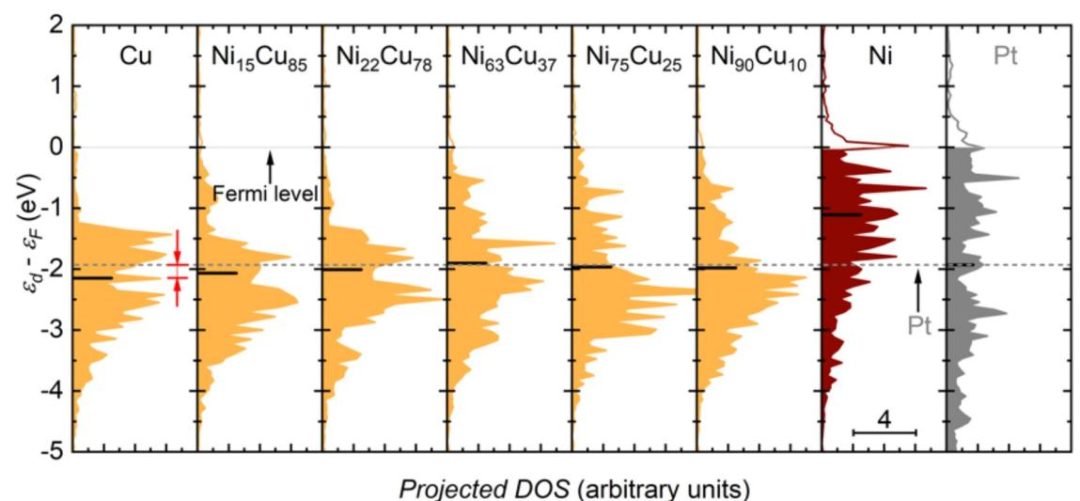
图4 具有不同比例的Ni原子的NiCu模型表面的投影d带态密度(DOS),由密度泛函理论(DFT)计算。Ɛd-Ɛf是d带谱(Ɛd)相对于费米能级(Ɛf)的能量。d带中心的位置由短黑水平条表示。灰色实线表示费米能级。灰色虚线表示Pt的d带中心。DOI:10.1021/jacs.9b12005
d带中心理论针对的是催化剂中活性位的金属原子,价带理论适用于非金属碳材料。价带理论选择活性原子的最高和最低轨道能之差(Ediff)作为活性描述符来预测非金属碳材料的催化活性。
电负性(χ)/平均电负性:反映元素或化合物吸引电子的能力。在合金或化合物中,平均电负性或组分电负性差异可影响电荷转移和吸附强度。高电负性金属通常增强电子转移效率,但需注意电负性差异的方向性(∆χ>0更优)以及与其他因素(如晶格氧空位、表面重构)的协同作用。
在催化剂设计中,优化电负性差异是提升电催化性能的有效策略之一。如图5,钴基钙钛矿的A位离子电负性(AIE)具有识别HER催化剂活性的能力。
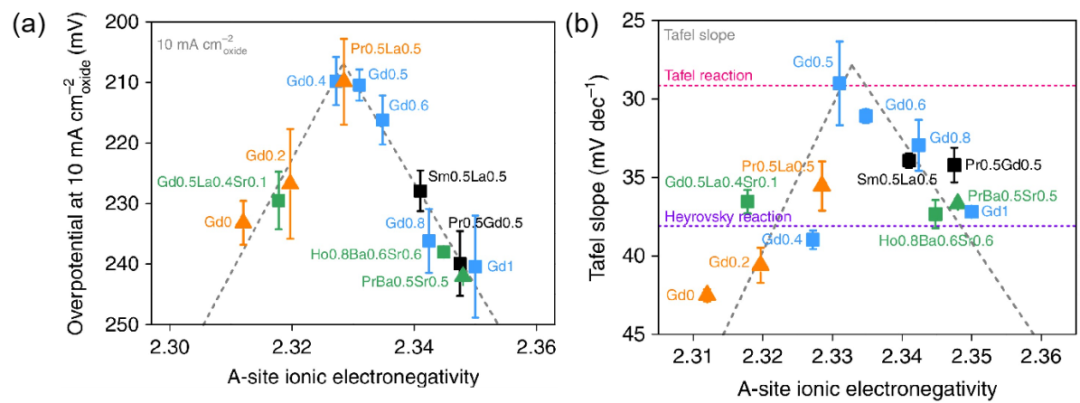
图5 单钙钛矿(三角形)和双钙钛矿(方型)在10mA cm
−2下HER(a)过电位和(b)Tafel斜率随A位点离子电负性的变化趋势。DOI:10.1038/s41467-019-11847-w
氧化态:金属中心的表观氧化态(尤其在氧化物中)影响其结合中间体的能力。例如,高价态金属(如Ni³⁺/Fe⁴⁺)常被认为对OER中的氧亲核攻击更有利。
电荷转移量:催化剂与吸附物种之间转移的电荷量(可通过计算获得),影响键的强度和活化程度。
功函数(Φ):材料费米能级到真空能级的能量差。影响电极/电解液界面的电子转移势垒和双电层结构,与催化活性相关,但作为单一描述符不如吸附能直接。
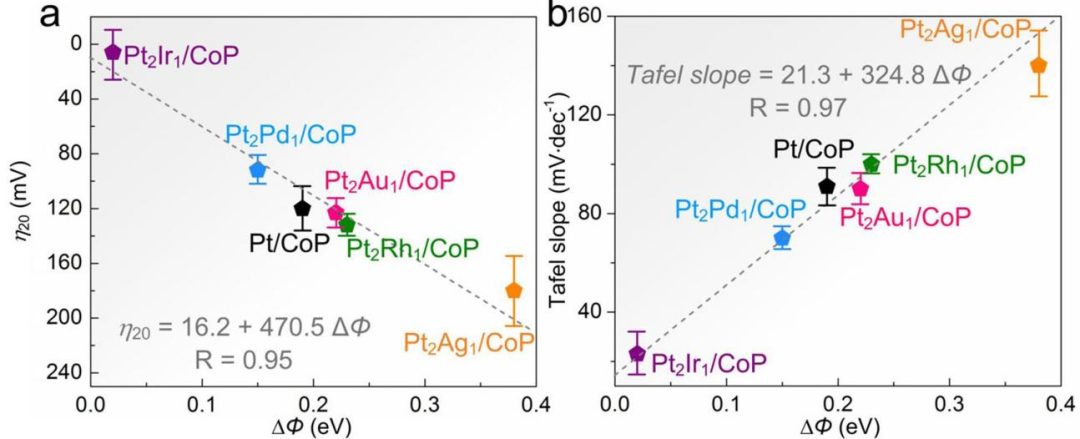
图6 各种PtM/CoP模型催化剂的本征HER活性及其ΔΦ的关系(a)电流密度为20mA cm
-2下的过电位(b)Tafel斜率。DOI:10.1038/s41467-021-23750-4
3、几何/结构描述符
配位数(CN):在中心原子(通常是金属离子或原子)周围,与其直接键合(通常通过配位键)的原子(配位原子)的数目。低配位数(如边、角原子)通常具有更强的吸附能力(不饱和配位),常表现出更高的本征活性(但也可能影响稳定性)。纳米颗粒、单原子催化剂的设计常围绕调控配位数进行。
M-N4中心是最常报道的活性位点,如Fe-N4、Co-N4分别用于氧还原反应(ORR)和析氧反应(HER),而低配位M-Nx构型显著促进HER活性。如图7,DFT计算表明,N-TM配位数的增加减弱了活性位点与H原子之间的相互作用,导致ΔGH*值增大。
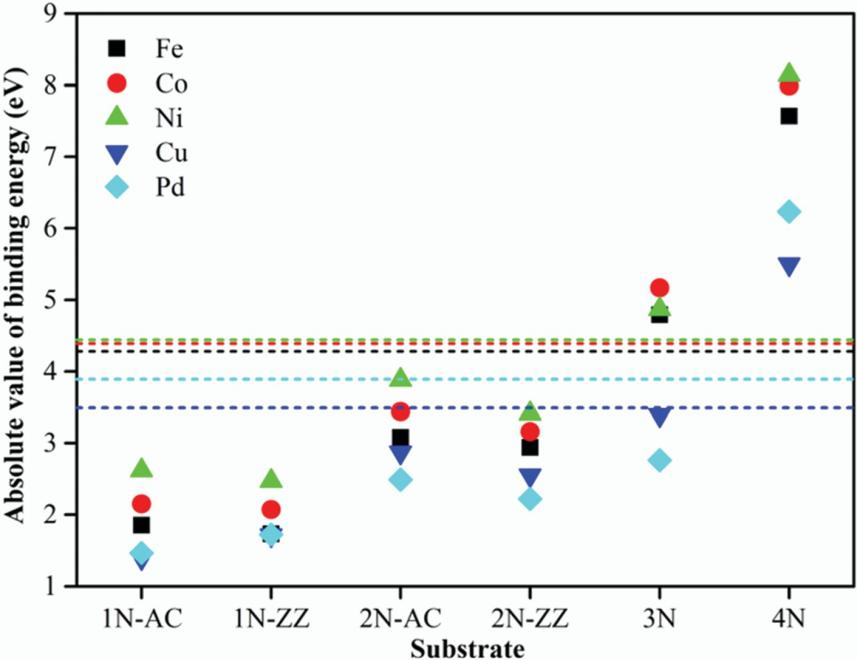
图7 结合能强度随着配位数N的增加而增大。DOI:10.1039/c8cp06755d
晶格应变/应力:由于晶格失配或尺寸效应引起的原子间距变化。压缩应变:通常使d带变宽,εd下移,减弱吸附。拉伸应变:通常使d带变窄,εd上移,增强吸附。广泛应用于核壳结构、异质结等催化剂设计。
位点对称性/局部原子排列:特定晶面或特定缺陷位点(空位、位错)具有不同的几何构型,显著影响吸附构型和强度。
4、材料本征性质描述符
电导率/电荷迁移率:影响催化剂本体和界面上的电子传输速率,对高电流密度下的性能至关重要(尤其在半导体氧化物中)。
稳定性指标:如溶解电位/腐蚀电位、相变能/形成能、表面能、比表面积等。其中,溶解电位/腐蚀电位可以表现催化剂电化学稳定性;相变能/形成能表现其热力学稳定性;表面能影响纳米结构的形貌和稳定性。
比表面积(SSA)/活性位点密度:虽然更多是外在性质,但高SSA和高活性位点密度是获得高总活性的基础(质量活性、比活性)。常与本征活性描述符结合使用。
重要考虑因素
如表1,中间体吸附自由能(ΔGads)及其衍生的理论过电位是当前最核心、最普适的热力学描述符。但是催化剂的性能往往受多个因素共同影响,单一描述符可能无法完全捕捉复杂行为,需要结合多个描述符(如吸附能+d带中心+配位数)或构建描述符矩阵。
表1 HER典型描述符。DOI:10.1002/advs.202200307

最佳描述符高度依赖于具体的电催化反应(HER,OER,ORR,CO2RR,NRR等)和反应路径。描述符主要源于理论计算(DFT),但其预测能力必须通过实验验证。
实验上测量真正的“本征活性”(如转换频率TOF)并与描述符关联是验证的关键。此外,吸附能描述符主要反映热力学趋势。真实的催化速率还受动力学(活化能垒、传质)影响。描述符通常更擅长预测趋势而非绝对速率。
电催化过程常在偏压下进行,催化剂表面结构、氧化态、吸附覆盖度可能发生动态变化(如OER中金属氧化物的表面重构),静态描述符有时需要谨慎使用或结合动态模拟。
总结
电催化剂描述符体系通过量化吸附自由能、电子结构(如d带中心)及几何参数(如配位数),构建了催化剂本征性质与性能的定量关联桥梁,为理性设计奠定基础。然而,单一描述符的局限性需依赖多参数协同解析,并需克服静态模型在动态反应环境中的适配挑战,最终通过实验验证实现精准预测与优化。