

负载型金属团簇催化剂是将具有特定结构和功能的金属团簇(由几个到几百个金属原子组成,尺寸通常在纳米尺度范围)分散负载于具有一定物理化学性质的载体材料表面或孔道内所形成的一类催化剂。
这类催化剂通过金属团簇与载体之间的相互作用,实现对金属团簇的结构调控、稳定化以及性能优化,从而在各类催化反应中发挥独特的催化作用,兼具金属团簇的高活性和选择性以及载体材料的稳定性和可调控性。


单金属负载型团簇催化剂:由单一金属元素组成的团簇负载于载体上,如负载型铂(Pt)团簇催化剂、负载型金(Au)团簇催化剂等。这类催化剂在一些特定的反应中展现出优异的催化性能,例如负载型 Pt 团簇催化剂在加氢反应中表现出较高的活性和选择性。
多金属负载型团簇催化剂:包含两种或两种以上金属元素组成的团簇负载于载体。通过不同金属之间的协同效应,可显著提升催化剂的性能。常见的如双金属(如 Pt – Ru、Au – Pd)负载型团簇催化剂,在一些复杂反应中,不同金属间的协同作用能够改变反应物分子的吸附和活化方式,提高反应效率和选择性 。
氧化物载体负载型金属团簇催化剂:以氧化铝(Al₂O₃)、二氧化钛(TiO₂)、二氧化硅(SiO₂)等氧化物为载体。氧化物载体具有丰富的表面羟基和多样的晶体结构,能与金属团簇发生多种相互作用,从而影响金属团簇的电子结构和分散状态,如 TiO₂负载的金属团簇催化剂在光催化反应中表现出色。
分子筛载体负载型金属团簇催化剂:分子筛具有规则的孔道结构和明确的酸性位点,将金属团簇负载于分子筛孔道内,可利用其独特的空间限制效应和择形催化特性,实现对特定反应的高效催化,如 ZSM – 5 分子筛负载的金属团簇催化剂在烃类转化反应中具有重要应用。
碳材料载体负载型金属团簇催化剂:以活性炭、碳纳米管、石墨烯等碳材料为载体。碳材料具有高比表面积、良好的导电性和化学稳定性,能为金属团簇提供稳定的支撑环境,还可通过表面改性等方式调控金属 – 载体相互作用,如石墨烯负载的金属团簇催化剂在电催化反应中展现出优异的性能 。
金属有机框架(MOF)及共价有机框架(COF)载体负载型金属团簇催化剂:MOF 和 COF 材料具有高比表面积、可调控的孔结构和丰富的化学活性位点,将金属团簇负载于此类材料上,可实现对金属团簇尺寸和分布的精准控制,同时材料本身的特性也有助于提升催化剂的活性和选择性。

结构优化与稳定性计算
通过密度泛函理论(DFT)计算,可以对负载型金属团簇催化剂的几何结构进行优化,确定其最稳定的构型。研究金属团簇在载体表面的吸附方式、吸附能,分析金属 – 载体之间的相互作用强度,从而评估催化剂的稳定性。
例如,计算不同金属团簇在氧化物载体表面的吸附能,判断哪种金属团簇更易稳定负载于该载体 。
电子结构分析
利用 DFT 计算研究负载型金属团簇催化剂的电子结构,包括电子云分布、能带结构、态密度等。通过分析电子结构,可以深入了解金属团簇与载体之间的电子转移情况,以及这种电子转移对催化剂活性位点电子性质的影响,进而揭示催化剂的活性和选择性来源。
反应机理研究
计算反应物分子在负载型金属团簇催化剂表面的吸附能、吸附构型,以及反应过程中各中间物种的能量和结构变化,确定反应的活化能和反应路径。
例如,在催化加氢反应中,通过 DFT 计算可以探究氢气分子和反应物分子在催化剂表面的吸附、解离以及加氢反应的具体步骤,为理解反应机理提供理论依据 。
催化剂性能预测
基于 DFT 计算结果,对负载型金属团簇催化剂的催化性能进行预测。通过改变金属种类、团簇尺寸、载体性质等因素,计算不同条件下催化剂的活性和选择性,为设计和优化高性能的负载型金属团簇催化剂提供理论指导,加速新型催化剂的研发进程。
以上系统梳理了负载型金属团簇催化剂的相关内容。若你还想对某些部分深入探讨,或有新的研究方向想加入,欢迎随时告诉我。
例1 : AM通过精准热切除原子级精确铜簇实现近红外光驱动的CO₂还原
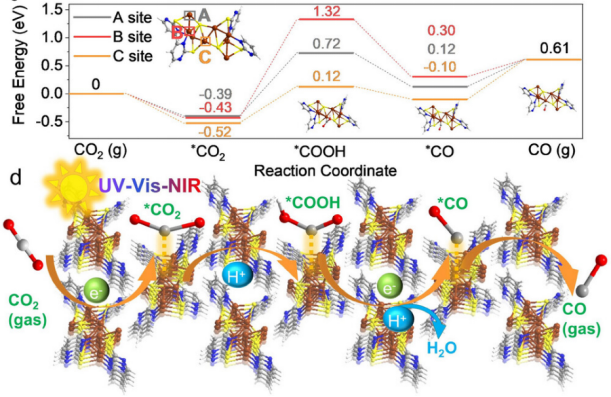
Adv. Mater. 2025, 2417747. https://doi.org/10.1002/adma.202417747
摘要:原子级精确铜簇因其明确的结构特性使其成为研究催化构效关系的理想模型,然而移除保护配体时易发生烧结导致结构破坏的问题长期制约其应用。
本研究采用混合配体策略成功合成铜-硫-氮簇[Cu8(StBu)4(PymS)4](Cu8SN),通过精确热切除技术选择性剥离弱配体叔丁基硫醇,诱导结构转变为新型稳定簇[Cu8(S)2(PymS)4](Cu8SN-T)。该转化过程中残留的桥连S2-形成具有近红外响应的不对称Cu-S活性位,使Cu8SN-T具备全光谱驱动CO2光还原能力,实现接近100%的CO选择性。
特别是在波长>780 nm的近红外光照射下,CO生成量可达42.5 μmol g-1。此项工作不仅首次报道了近红外光响应的铜簇催化剂,更开创了通过精准热解策略调控金属团簇结构的新范式,为开发新型功能化金属团簇材料提供了全新思路。
例2:Nat. Commun.:块体Cu2O的表面工程用于高效电合成尿素
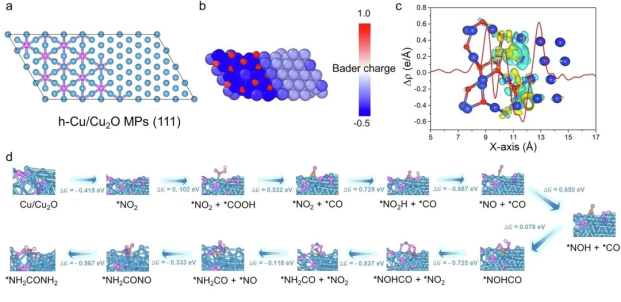
Nat. Commun. 2025, 16, 1-13. https://doi.org/10.1038/s41467-025-57708-7.
摘要:电化学尿素合成近年来因其高效节能特性成为备受关注的绿色合成途径,但如何同步实现高生产速率与高法拉第效率仍是关键挑战。本研究通过块体Cu2O的原位电化学工程构建Cu/Cu2O微颗粒异质界面,在温和条件(-0.3 V vs. RHE,常温常压)下实现了CO2与NO3⁻的高效共还原,成功打通低能耗电化学C-N偶联路径。该催化剂展现出优异的尿素合成性能:产率达632.1 μg h-1 mgcat-1,法拉第效率达42.3%,为目前报道的最高水平之一。通过原位同步辐射傅里叶变换红外光谱与理论计算联合分析,揭示了异质界面处NOH与CO中间体的动态耦合机制——界面诱导的电子结构调控有效降低了C-N键形成能垒。
这项工作不仅为电催化尿素合成提供了高效催化剂设计范例,更为理解C-N偶联反应机制提供了原子级见解,推动绿色氮肥合成技术的创新发展。
例3:Angew基于CaCO3上原位溶解的Cu纳米团簇动态稳定的Cu0Cuδ+对位点,用于高效电还原CO2
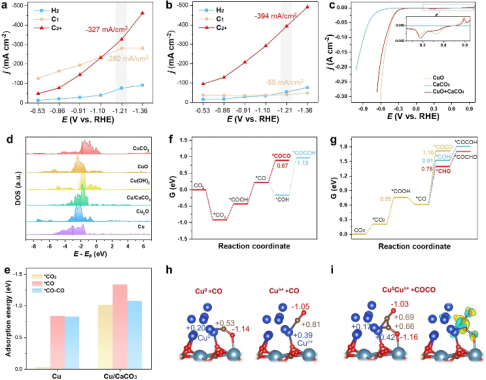
Angew.Chem.Int.Ed.2025,e202421860. https://doi.org/10.1002/anie.202421860.
摘要:铜基催化剂是在二氧化碳电还原(CO2RR)过程中生成多碳产物(C2+)的首选催化剂,其中Cu0Cuδ+对位点是C-C偶联的协同热点。保持它们的动态稳定性需要精确控制电子亲和力和阴离子空位形成能,这给我们带来了巨大的挑战。
在这里,作者提出了一种原位重构策略,在铜纳米团簇和CaCO₃纳米球(Cu/CaCO₃)的界面上创建动态稳定的Cu0Cu0.18+OCa。原位XAFS分析证实了CO2RR过程中Cuδ+的低价态。
DFT计算表明,纳米团簇的大小来自金属–载体相互作用和Cu-Cu结合能之间的平衡,而富含界面Cuδ+位点的动态稳定性则归因于它们低的电子亲和力和高的CO32-空位形成能,这些因素共同导致了还原性降低。
在部分电流密度为393 mA cm-2时,转化后的Cu/CaCO₃表现出83.7%的C2+法拉第效率,这是因为异质铜位点吸附了电负性不同的*CO,从而降低C-C耦合能垒。研究结果表明,不溶性碳酸盐是Cu0Cuδ+位点的有效阴离子配对,突出了原位重组策略在产生高密度动态稳定的Cu0Cuδ+配对位点方面的有效性。
例4:将CeO2转化成二维团簇可以增强催化活性
本研究利用像像差校正/高角度环形暗场扫描透射电子显微镜(AC-STEM/HAADF-STEM)、原位红外光谱、X射线光电子能谱(XPS)、扩展X射线吸收精细结构(EXAFS)、密度泛函理论(DFT)建模和催化性能测试等先进工具,对负载型铈基纳米材料提供了新的原子层面的认识,结果表明,大约一个原子层厚度的CeₓOᵧ区域具有卓越的氧迁移性,并且在与工业相关的条件下,其催化活性得到了显著提高。
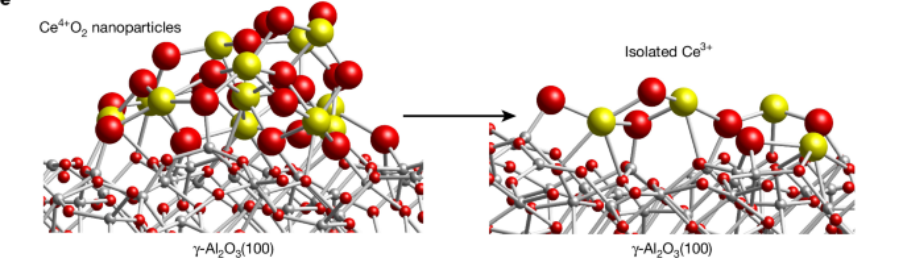
Nature (2025): 1-7.
例5:原子精确纳米团簇电催化析氢
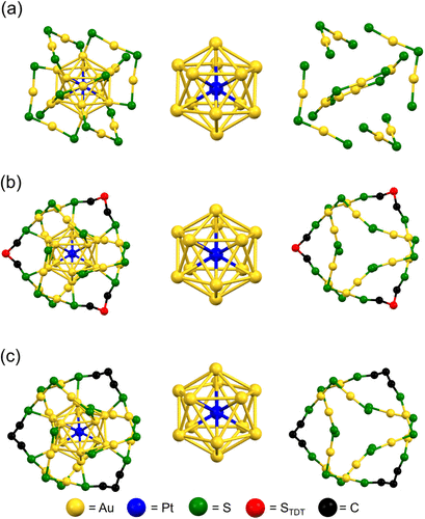
J. Am. Chem. Soc. 2024, 146, 43, 29684-29693. https://doi.org/10.1021/jacs.4c10868.
摘要:[Au24Pt(C6)18]0(C6 = 1-己硫醇)在促进电催化析氢反应(HER)方面的活性是商用Pt纳米粒子的两倍,因此作为具有良好几何结构的新型HER催化剂而备受关注。
在这项研究中,作者成功合成了两种新的Au−Pt合金纳米团簇,即[Au24Pt(TBBT)12(TDT)3]0(TBBT = 4-叔丁基苯硫酚;TDT = 硫代二硫醇)和[Au24Pt(TBBT)12(PDT)3]0(PDT = 1,3-丙二硫醇),方法是将[Au24Pt(PET)18]0(PET = 2-苯基乙硫醇)的配体全部换成单硫醇或二硫醇。
虽然[Au24Pt(TBBT)12(TDT)3]0是偶然合成的,但随后通过选择适当的反应条件以及硫醇和二硫醇配体的最佳组合,得到了类似的团簇化合物[Au24Pt(TBBT)12(PDT)3]0。单晶X射线衍射分析表明,[Au24Pt(TBBT)12(TDT)3]0和[Au24Pt(TBBT)12(PDT)3]0中-Au(I)-SR-Au(I)-主链的长度和取向与[Au24Pt(C6)18]0中的不同、[Au24Pt(TBBT)12(TDT)3]0和[Au24Pt(TBBT)12(PDT)3]0的几何结构、电子结构以及HER活性都反映了这些细微的差异。
因此,产物[Au24Pt(TBBT)12(TDT)3]0和[Au24Pt(TBBT)12(PDT)3]0的HER活性分别是[Au24Pt(C6)18]0和[Au24Pt(TBBT)18]0的3.5倍和4.9倍。