

Q1:朱老师,算d带中心是积分到费米能级还是全域积分?
A:全域
Q2:朱老师,解释chco在表面上的吸附能大于ch2co,可以用表面向chco的电荷转移大于ch2co解释吗?
A:可以用电子转移的,还有差分电荷
Q3:朱老师,我建了一个NiO和Ni的界面模型,在结构优化的时候报错啦,这个咋解决呀?
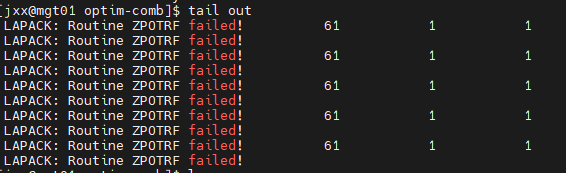

A:调整下amix
Q4:朱老师您好,我在计算CO2RR台阶图时,涉及到CC偶联,如CHOCHO中间体,想问下您在进行质量守恒时,可以用H2O和H2分子进行加减守恒吗?还是说只要偶联成C2以后,都需要直接算单独的自由中间体的能量,然后用总能量减去slab 再减去单独中间体即可?
A:用h2o和h2去做到质量守恒
Q5:朱老师您好,我在计算过渡态的时候,一直出现下面的报错。
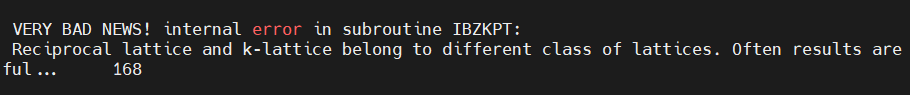
A:这个不是错,可以接着算
Q6:朱老师,如果自己积分COHP的话 是只积分成键那一部分么 就是-COHP为正值 费米能级以下部分?
A:不是,全部,成键反键都要
Q7:朱老师,功函数的大小与氧吸附强度可以关联吗 具体关系是什么呢?
A:有关系,但一般不是线性
Q8:朱老师,如果我固定其它原子不弛豫、只有特定的原子弛豫,那还有必要设置EDIFF和EDIFFG吗?如果有必要,那此时它们针对的对象(即EDIFF和EDIFFG此时的物理含义)是什么呢?
A:需要设置,针对整体能量,和原子受力
Q9:朱老师,我还想咨询一下NELECT参数的设置。文献上说:把晶体结构Na3Zr2Si2PO12移除一个钠离子创造空位(one Na+-ion was removed to create Na vacancy),然后用背景电荷平衡(The defect charge was compensated by a uniform background charge to retain the charge neutrality)。
那么,请问我这个NELECT该怎么设置呢,是设置原体系Na3Zr2Si2PO12的电子数-10(一个钠离子的电子数),还是原体系Na3Zr2Si2PO12的电子数-11(一个钠原子的电子数)呢?
A:去掉na原子,不设置其他,这是0价,加上一个电子nelect在0基础上加1就是-1价
Q10:朱老师你好,我在vasp里面用DFPT计算介电常数,IBRION=8, ISYM = 0, 且已经打开LOPTICS = .T.,LEPSILON = .T.,但是输出没有介电张量的IONIC contribution部分,请问是什么原因?
A:dfpt不用loptics,也不用isym,去掉这两个参数试试