在催化领域,理论计算,特别是基于密度泛函理论(DFT)的方法,起着至关重要的作用。它可以从原子和电子层面揭示催化反应的本质,帮助预测材料的吸附性能、反应路径以及能垒高低。
例如,通过计算反应中间体的吸附能,可以判断催化剂对特定反应物的亲和力,进而推测催化活性。同时,理论计算还能分析催化剂表面的电子结构变化,如带隙、态密度分布等,指导催化剂的合理设计。
此外,结合过渡态搜索(如NEB方法)可以确定反应速率决定步骤,为优化反应动力学提供依据。随着计算资源的提升和机器学习的引入,理论计算在催化材料筛选、新反应机制预测等方面的应用越来越广泛,成为催化研究不可或缺的重要工具。
在催化研究领域,密度泛函理论(DFT)堪称绝对的大哥大,几乎成为每一篇高水平论文中的必备工具。DFT能以相对可控的计算量,在原子尺度上准确预测材料的电子结构、吸附能、反应能垒等关键参数,为理解催化机理提供直接证据。
例如,黄洪伟教授团队利用DFT系统研究了压电材料的带隙和电子结构,发现材料内部的极化电场可以调控表面反应过程,显著增强催化活性。这种微观层面的洞察不仅帮助设计新型催化剂,也指导了实验条件的优化。
DFT的应用范围极广,从简单气相分子反应,到复杂固体表面催化,甚至涉及界面、缺陷、掺杂等多因素影响,使其成为理论催化不可替代的基石工具。
npjComput Mater 7, 137 (2021). https://doi.org/10.1038/s41524-021-00605-6
另一个越来越受关注的工具是恒电势计算,尤其在电催化领域非常重要。传统DFT计算通常在电荷中性条件下进行,但实际电催化反应中,电极表面存在电势和表面电荷。
为了更真实地模拟界面反应,研究者利用VASP等软件平台,通过调节表面电荷密度、引入背景电荷或显式溶剂模型,进行恒电势下的DFT计算。这样可以更准确地捕捉电极表面吸附物种的能量变化、反应路径以及电场对吸附构型的调制,极大提高了电催化机理研究的可靠性。
随着理论方法不断进步,这些多尺度、多方法的结合正在推动催化科学向着更精准预测和理性设计方向迈进。
除了DFT之外,催化研究中还有几个非常重要的辅助“小弟”,其中之一是分子动力学(MD)模拟。MD通过数值积分牛顿运动方程,能动态追踪原子和分子的运动轨迹,适合研究催化剂表面的重构、扩散以及高温稳定性等现象。
例如,南方科技大学与新加坡国立大学的研究者合作,采用经典MD模拟双金属纳米颗粒的表面偏析行为,在高达873K甚至1773K的条件下,观察不同元素如何在高温下迁移并重新分布。这种动态过程是DFT静态计算无法直接捕捉的,对理解催化剂的寿命、热稳定性与活性演变至关重要。
近年来,机器学习(ML)与DFT的结合成为催化研究的一大新趋势。传统DFT筛选新催化剂往往计算量庞大,周期长,而韩国科学技术研究院的研究者开发了基于机器学习的预测框架,结合d带中心理论与火山图(volcano plot)模型,大幅加速了材料筛选流程。
通过训练模型识别结构–性能关系,他们能在成千上万个候选材料中快速挑选出潜力催化剂,大大缩短了从理论设计到实验验证的时间。这种方法不仅提升了研究效率,也拓展了催化剂设计的可能性空间。
npjComput Mater 7, 137 (2021). https://doi.org/10.1038/s41524-021-00605-6
这些方法的核心思想都是“从头算”,不用实验数据,直接基于量子力学。比如DFT的计算流程一般是:建模型(比如表面、掺杂)→优化结构→算能量→分析电子态(能带、态密度)→找过渡态→算反应路径。
理论计算在催化领域的应用范围极其广泛,已经成为指导实验、揭示机理和开发新材料的重要工具。首先,理论计算可以用于预测材料的性能。
例如,厦门大学团队通过密度泛函理论(DFT)研究了铂(Pt)表面氨氧化反应的机制,系统计算了不同电位下的反应路径,发现高电位时反应偏向O-S机制,而低电位下则转为另一种路径[26]。
这种从原子尺度获得的机理信息,直接指导了实验在不同操作条件下催化剂的选择与优化,有效提高了催化反应的效率和选择性。因此,理论计算不仅是机理解释工具,更是实验设计的重要前哨。
另一个关键应用是优化吸附能。吸附能直接关系到催化剂表面对反应物的活化能力,是决定催化活性的核心参数之一。
以单原子催化剂为例,通过DFT计算可以精确得到反应物(如CO)在金属位点(如Ru)上的吸附能量。如果吸附太强,反应物会在表面滞留,堵塞活性位点,导致催化剂中毒;如果吸附太弱,则无法有效活化反应物。
通过理论优化吸附能,可以为单原子催化剂的设计提供重要指导,寻找到既能高效吸附又易于反应产物脱附的最佳平衡点。
理论计算还在寻找反应过渡态方面发挥重要作用。反应速率通常由过渡态的能垒高度决定,找出关键过渡态对理解反应速率控制步骤至关重要。
比如,在研究氢分子(H₂)在铝(Al)表面的解离过程时,研究者通过NEB(弹性带)方法计算了整个反应路径的能量变化,发现掺杂钛(Ti)元素可以显著降低解离能垒,从而加速吸氢过程。这种基于能垒调控的设计策略,为开发高效储氢材料和催化剂提供了理论依据。
最后,理论计算在新材料设计中同样不可或缺。对于如高熵合金催化剂这类组分极其丰富的体系,实验筛选几乎不可能全面覆盖,而DFT可以高效计算不同组分组合的合金形成能和表面偏析能,快速筛选出稳定性好、催化潜力高的候选材料。
通过这种“计算先行,实验验证”的策略,极大提高了新材料开发的效率与成功率。总体来看,理论计算已成为催化科学中不可替代的核心支撑力量。
催化论文里的DFT图五花八门,但常见的有以下几类:
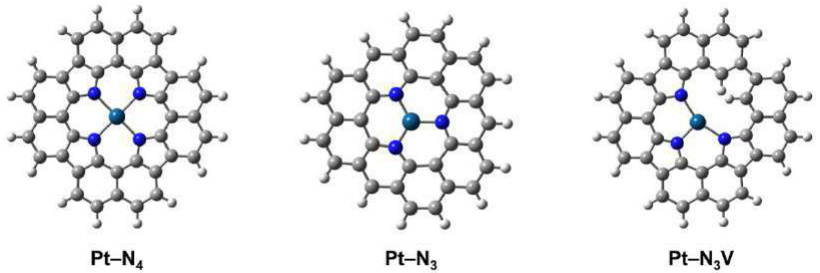
首先是活性位点模型图。下图直观展示了催化剂表面原子的排列情况和具体的活性位点分布,不同颜色代表不同的元素,比如蓝色代表Pt,绿色代表N等,箭头则标示出反应物吸附的位置。
分析这类图时,重点是观察活性位点的几何构型(如四面体、平面结构)以及周围配位环境(如典型的Pt-N₄结构)。这些因素会直接影响催化剂对反应物的吸附能力、反应中间体的稳定性以及最终的催化活性和反应选择性,因此是理解催化性能差异的基础。
Graphite Conjugated Nickel Phthalocyanine for Efficient CO2 Electrodreduction and Zn-CO2 Batteries. Jingwei Han et al.[2024]
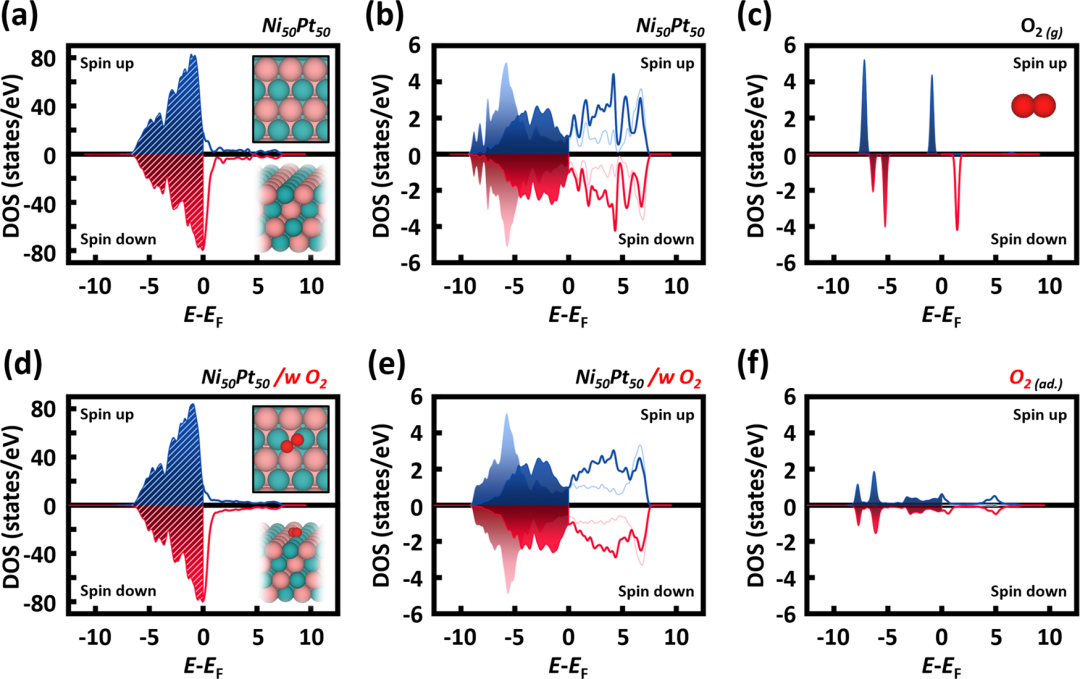
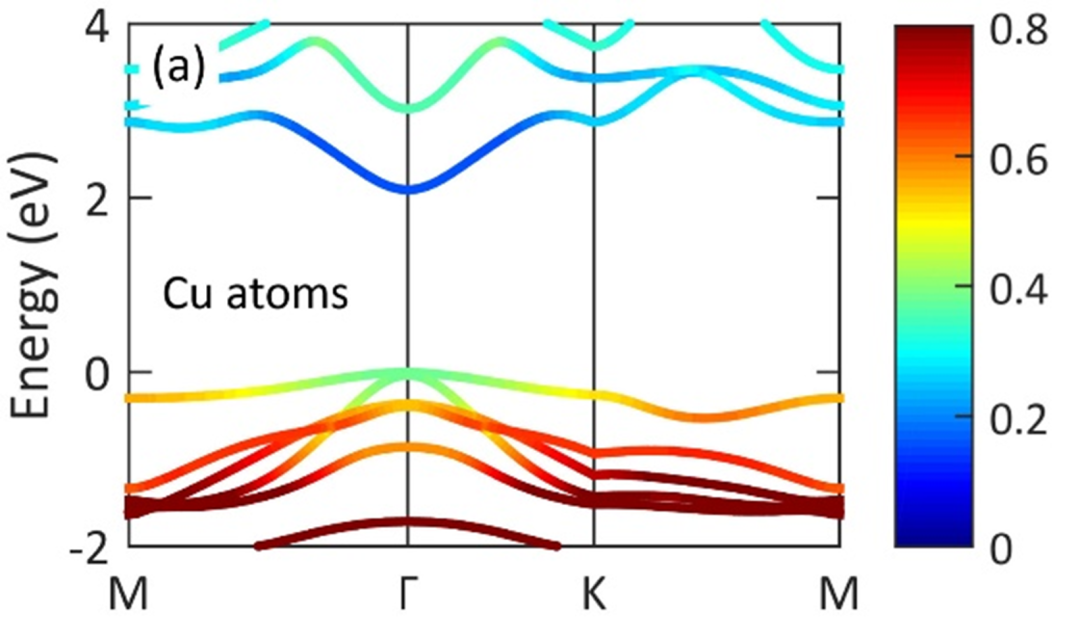
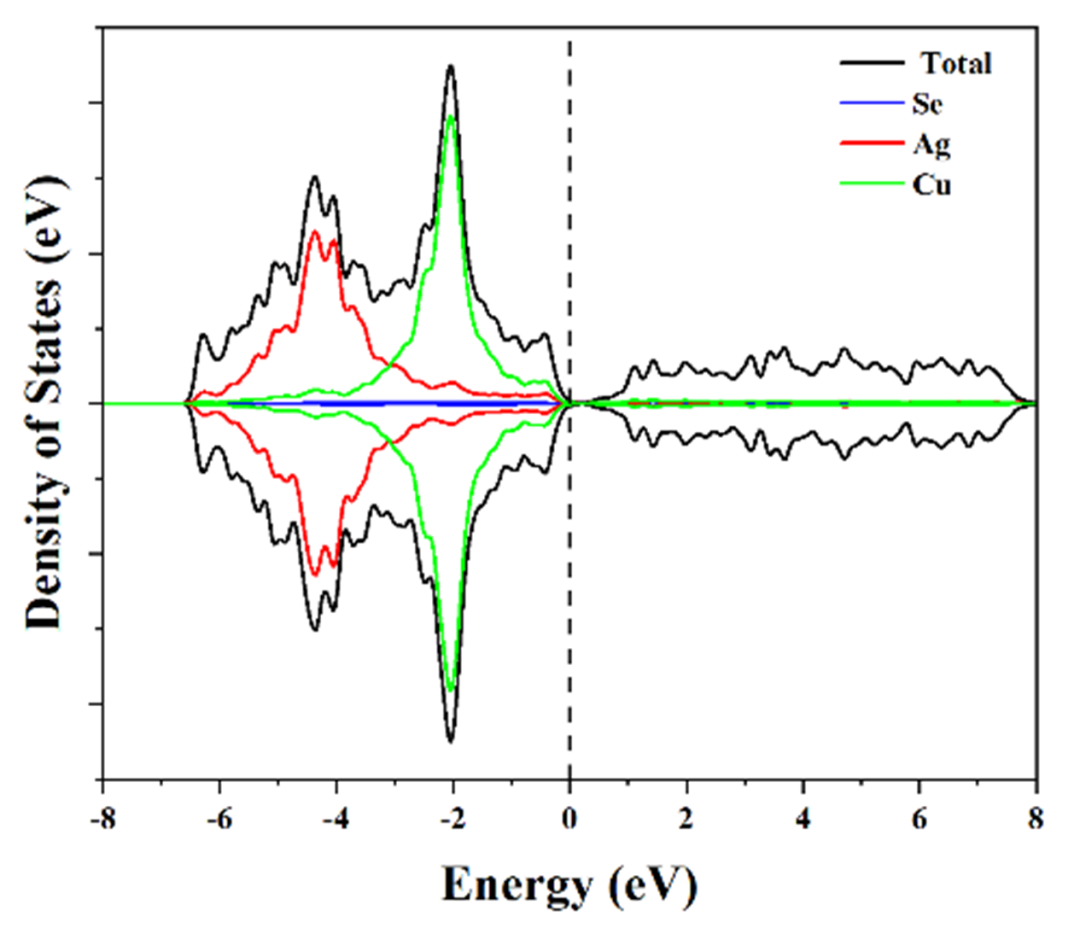
其次是能带结构图和态密度(DOS)图。能带图展示了材料在电子结构上的导电性特征,带隙宽度直接决定了材料的导电或光吸收性能。而态密度图则更细致地分解到各个原子轨道的贡献,例如分析d轨道的分布情况,尤其是d带中心的位置。
分析这类图时,需要注意:导电性好的材料(如带隙窄或无带隙的金属)更适合电催化应用,而带隙适中的半导体材料则更适合光催化应用。同时,d带中心上移通常意味着与反应物(如CO分子)的吸附更强,这可以用来调控催化剂的选择性和活性。
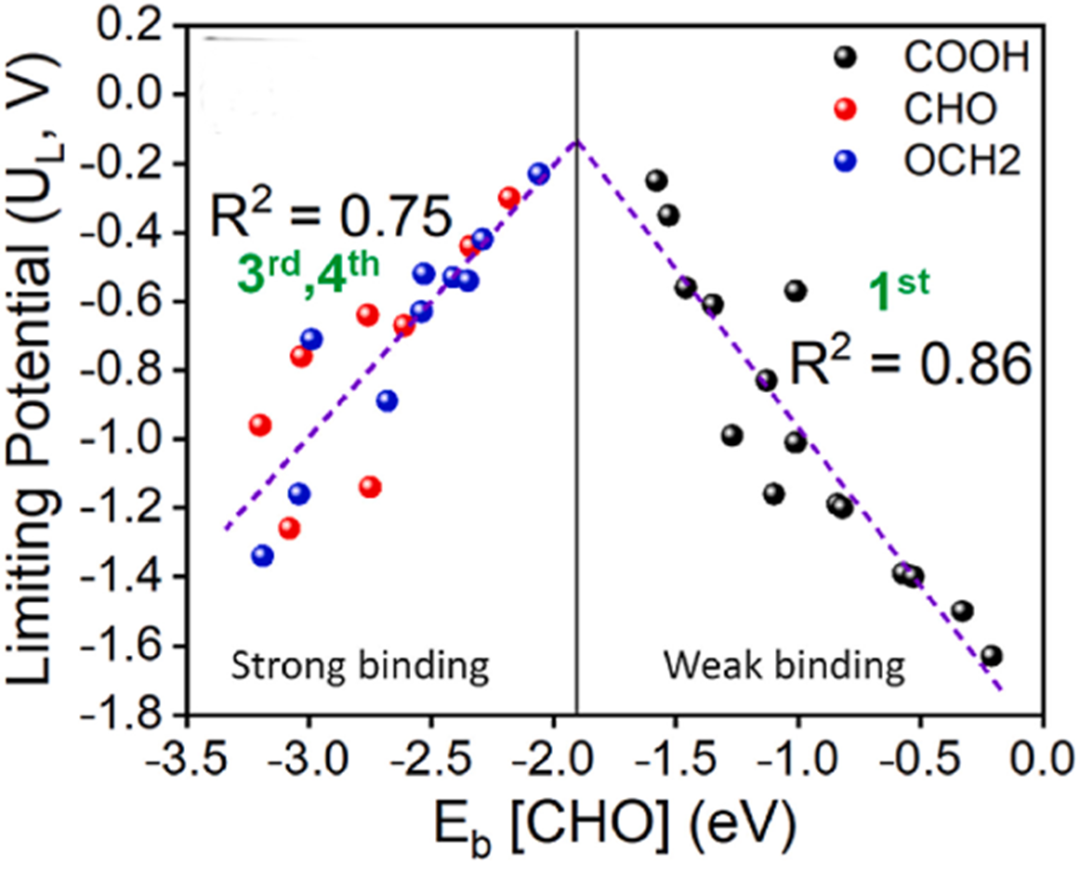
第三种是自由能变化图,这在解析电催化反应机制时尤为常见(针对OER反应)。自由能图展示了反应过程中每一步的自由能变化(ΔG),每一个高峰代表一个能垒大的反应步骤。分析时应重点关注ΔG最高的那一步,因为它通常是反应的决速步骤。
例如,如果在第三步出现了ΔG=1.8 eV的高能垒,那么这一步就极大地限制了整体反应速率。改进催化剂设计时的策略就是降低这一步的能垒,从而加速整体反应动力学,提高效率。
再者是非常经典的火山图。火山图通常以吸附能为横轴,催化活性为纵轴,形状呈火山状。比如在析氢反应(HER)中,吸附能过强会导致反应物粘在表面不易脱附,过弱又无法有效吸附反应物,只有适中时催化活性最佳。
因此,在火山顶附近的材料性能最优,比如Pt就是HER反应中位于火山顶附近的理想催化剂。通过火山图可以直观快速地筛选出潜力催化剂,大大提高研究效率。
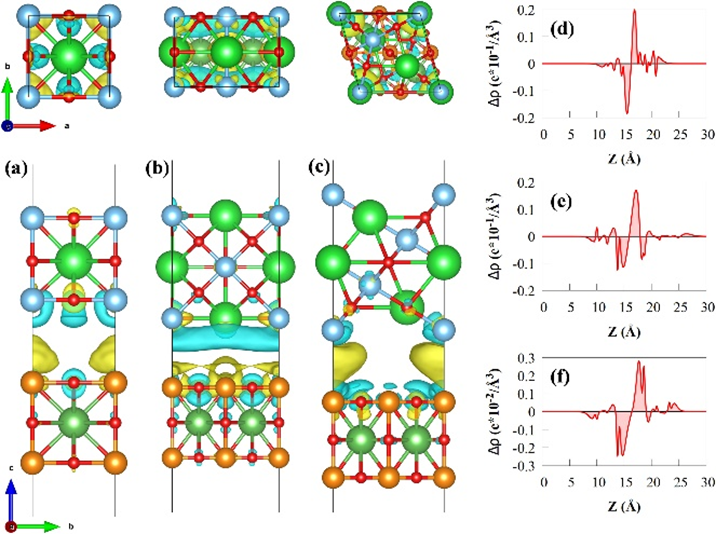
最后一种是电荷密度差图,用于分析反应过程中的电荷转移情况。图中通常红色区域表示电子积累,蓝色区域表示电子流失。这类图可以揭示催化剂表面与反应物之间的键合机制。
例如,在CO分子吸附到金属表面时,金属原子会向CO的反键轨道注入电子,导致C-O键的削弱,从而促进分子的活化。通过观察电荷密度差分布,可以深入理解吸附和反应机制,为优化催化性能提供重要依据。
在该文中,理论计算部分主要运用了密度泛函理论(DFT)计算,从多个关键方面深入探究了Ru/CeO2光催化剂的性能与反应机制。
在对 Ru 原子的化学状态及催化剂电子结构的研究中,通过构建CeO2 (111) 晶面的 Ru 单原子替代模型,计算发现引入 Ru 单原子后,Ce – O 键伸长,Ru 单原子的 Bader 电荷为正,发生电子从 Ru 到CeO2的重新分布,其配位环境也得到确定,为解释催化剂的光催化性能提供了结构基础。
同时,DFT 计算结合态密度(DOS)分析表明,引入 Ru 单原子使态密度整体向低能量方向移动,Ru 3d 电子与 O 2p 电子有效重叠,Ce 的 d 带中心负移,更易失电子参与光还原反应,这对光催化反应的进行具有重要意义。
https://doi.org/10.1021/acsnano.3c12001
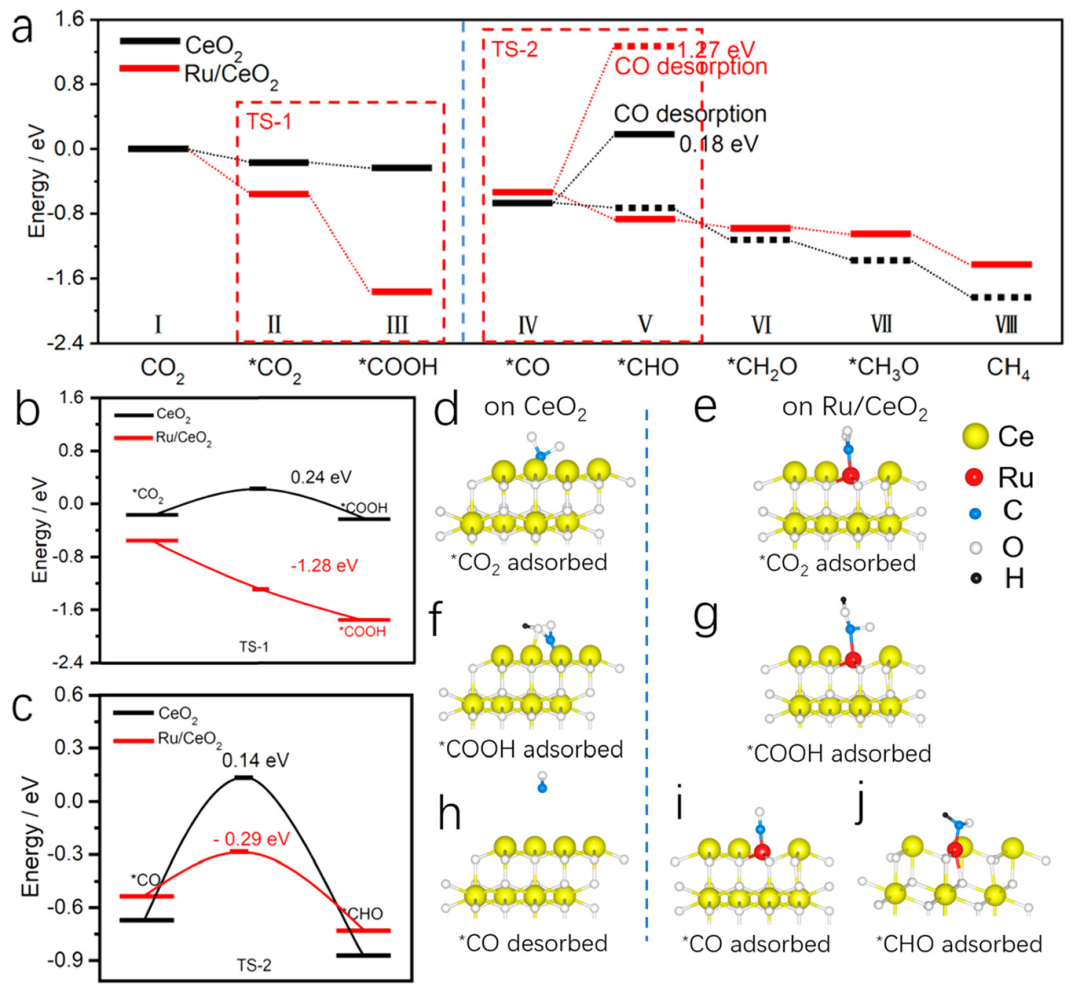
在CO2还原反应路径及中间体吸附能的研究上,DFT 计算表明,Ru/CeO2对CO2的吸附能比CeO2更高,这使得CO2在Ru/CeO2表面更易被吸附和活化。
在反应过程中,∗COOH在Ru/CeO2上的形成能垒远低于CeO2,且∗CO在Ru/CeO2上的脱附能是CeO2的 7 倍,这使得∗CO在Ru/CeO2上更稳定,更有利于进一步加氢生成CH4。
这些结果与原位 FT – IR 分析相互印证,共同揭示了Ru/CeO2光催化剂上CO2光还原生成CH4的反应机制。
理论计算在催化领域就像“数字显微镜”,能看清原子尺度的细节。但它也有局限,比如DFT对强关联体系(如某些氧化物)算不准,这时候得用DFT+U或杂化泛函修正。
未来,机器学习加速计算、高通量筛选会成为趋势,比如Meta AI的OC22数据库用9百万个DFT数据训练模型,预测吸附能比传统DFT快1亿倍!
总之,理论计算和实验就像左右手,缺一不可。搞催化的同学,赶紧学起来吧!
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!