

目的:介绍Cantera模块在表面多相催化中的应用,并说明得到表面体系反应机理的过程。
所用模块:Materials Visualizer、Cantera、DMol3(仅使用分析功能,可选)
前提条件:无
背景
多相催化是化学中最基本的过程之一。要理解它,不仅需要从原子水平上理解潜在的反应速率,而且需要一种可以将表面和气相反应机制集成的研究工具。Materials Studio的Cantera结合DMol3模块提供了此类工具。
在本教程中,将使用DMol3进行一系列密度泛函理论计算,并在Cantera模块中使用这些结果。使用了一个近似模型来研究在RuO2(110)表面上CO的催化氧化(Reuter et al., 2004)。将着重于研究吸附和脱附过程,但忽略表面上的扩散过程。
该表面上的反应发生在钌原子上,钌原子排列成分离的行。表面上有两种不同类型的钌原子(称为桥位和cus位),导致产生两组吸附位点和反应物和产物相似的众多反应。我们将在Materials Studio Cantera模块中详细讨论如何处理这种情况。
在本教程开始时,将在Cantera数据表中使用部分完整的反应机制,并学习如何生成表面反应机制。然后,将使用此机制在两个不同的反应模型上执行计算。
介绍
本教程包括如下部分:
- 开始
- 使用反应编辑器Reaction Editor修改Cantera输入数据
- 设置Cantera表面平衡计算参数
- 设置Cantera推流式反应器计算参数
注意:为了确保您可以完全按照预期的方式学习本教程,您应该使用“设置管理器(Settings Organizer)”对话框确保项目中所有参数都设置为BIOVIA的默认值。
1、开始
首先启动Materials Studio并创建一个新项目。
打开New Project对话框,输入surface作为项目名,单击OK按钮。
新项目将以surface为项目名显示于Project Explorer中。
单击“导入Import”按钮


打开 Import Document对话框。导航到ExamplesCanteraSurface Reactions文件夹并导入RuO2_surface_incomplete.std数据表文档。
单击Modules工具栏上的Cantera按钮

选择Reaction Editor或从菜单栏中选择Modules | Cantera | Reaction Editor。从Study table下拉列表中选择RuO2_surface_incomplete.std。
选择数据表的Reference表单,双击Structure列中的O2Ru_surface单元。
将显示氧化钌的表面模型。
在RuO2(110)表面最顶层,有两排不同的非等效钌原子。五配位钌原子位于氧原子正上方,并与顶层的另外四个O原子结合。将此吸附位置称为cus位置。四配位钌原子与顶层的两个氧原子和下层的两个氧原子结合。称之为桥位。在每个表面单位晶胞中,均含有一个这两种类型的吸附位置。在本教程中,Materials Studio和Cantera可以区别这两种不同的吸附位置,从而在反应机制和模拟计算中保留正确的表面结构。
打开Cantera Reaction Editor对话框时,可自动解释数据表详细信息视图的内容。在Reference species部分中,得到了8.41×10-10 mol/cm2的表面位置密度Surface site density,该密度是根据体系初级胞的表面积计算得出的。在该情况下,每个表面单位晶胞有两个吸附位点。这意味着RuO2_surface_incomplete.std数据表Reference选项卡上的表面位置密度Surface site density应为Reaction Editor自动计算值的两倍。
选择数据表的Reference表单,定位至表面位置密度(mol/cm/cm)Surface site density (mol/cm/cm)列。检查此列中的值是否是Cantera Reaction Editor对话框中的值的两倍。
在Cantera Reaction Editor对话框的Reactions and structures部分中,将该体系识别为衬底,并命名为桥位bridge。数据表的化合物选项卡中实际上包含相同结构的两个副本(桥位和cus位),对应于RuO2表面上的两个可能吸附位置。选择这两种结构中的一种作为该化合物的建议名称。
注:Reactions and structures列表中的Add列值为No,这是因为该化合物已存在于数据表中。
关闭数据表详细信息视图。
2、使用Reaction Editor修改Cantera输入数据
数据表缺少反应:
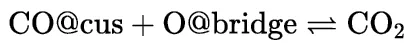
打开Import Document对话框并导入文件COcus+Obridge.xod。
导入的收集文档包含缺少的反应。首先需要计算速率常数。
单击模块工具栏上的DMol3按钮 ,然后选择Analysis或从菜单栏中选择Modules |DMol3 | Analysis。
,然后选择Analysis或从菜单栏中选择Modules |DMol3 | Analysis。
从列表中选择“反应动力学Reaction kinetics”。
将温度范围temperature range设置为从From:200 K至To:6000 K。
单击Calculate,并关闭对话框。
创建了一个名为COcus+Obridge.std的数据表,其中包含有关反应的信息。
检查新的研究表,确保反应速率参数的拟合值合理。例如,修正的Arrhenius方程的拟合活化能Ea应为正值,且具有正确的标度,接近20 kcal/mol。
在Graphs选项卡上,选择F-H、K和L列。单击Quick Plot按钮 以获取该反应的计算和拟合速率图。
以获取该反应的计算和拟合速率图。
选择数据表中的Structures选项卡,并检查Structures列中的不同文档。
第一行包含原始收集文件的副本,第二行和第三行包含单独的反应物Reactant和产物Product结构。Cantera Reaction Editor无法直接导入这两个结构:反应物是一个具有两个运动基团的表面,而产物是表面上方的气相分子,都需要对化合物进行单独计算。
提示:在某些情况下,可能只有过渡态、反应物和产物为单独的3D原子文档。按照DMol3部分联机帮助中有关如何从原子数据创建收集文档的说明进行操作,这些数据仍然可以导入到Cantera数据表中。
在Cantera Reaction Editor对话框中查看Reactions and structures列表。
其中包含反应所需结构。列表中的Add列值为NA,因此不能将化合物作为反应所需结构导入。
注:吸附的一氧化碳分子命名为CO@bridge,对应于Cantera Reaction Editor对话框在数据表中所找到的结构。Cantera Reaction Editor对话框可以检测表面上的分子,但无法区分不同的吸附位置。
在导入反应之前,必须手动修改。
在Cantera Reaction Editor对话框的Reactions and structures列表中,将一氧化碳化合物的名称Name从更改为CO@bridge到CO@cus.
单击Import按钮。
Cantera将提示错误信息。
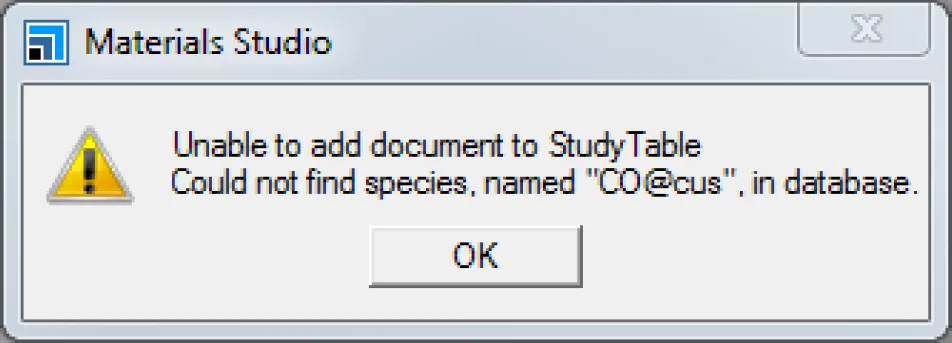
 ,打开Import Document对话框并导入文件CO@RuO2_110_cus.xsd。
,打开Import Document对话框并导入文件CO@RuO2_110_cus.xsd。