

环丙烷广泛存在于天然产物和生物活性分子中,其合成方法一直是有机化学的研究热点。传统方法主要依赖于金属催化剂(如铜、铑、钌配合物),虽已实现高对映选择性,但存在区域选择性差(尤其对多烯烃底物)等局限性。有机催化虽可避免金属使用,但仅适用于吸电子基团活化的烯烃,对电子中性烯烃的催化尚无有效方法。光氧化还原催化通过单电子转移生成自由基阳离子中间体,为烯烃活化提供了新思路,但如何结合手性抗衡阴离子实现高对映选择性仍是挑战。
基于此,德国马克斯·普朗克煤炭研究所Benjamin List教授和Chandra Kanta De博士等人设计了一种新型催化反应,利用光激发的阳离子有机催化剂激活烯烃为自由基阳离子中间体,并与手性反离子形成离子对,进而与重氮烷烃发生对映选择性碳-碳键的形成。该方法适用于多种烯烃,解决了多烯烃底物的区域选择性问题。该研究以“Organocatalytic regio- and stereoselective cyclopropanation of olefins” 为题,发表在《Nature Catalysis》期刊上。值得注意的是,本文第一作者为南京大学校友Zhu Chendan。



Benjamin List,2021年诺贝尔化学奖得主,德国马克斯·普朗克煤炭研究所(Max-Planck-Institut für Kohlenforschung)所长、德国科学院院士。利斯特 1968 年出生于德国法兰克福,1997年在法兰克福大学(University Frankfurt)获得博士学位。1999年在美国斯克利普斯研究所(Scripps Research Institute)任副教授,随后加入马克斯·普朗克煤炭研究所。利斯特目前还担任德国科隆大学荣誉教授。他曾于2009年获得汤森路透引文桂冠奖(Thomson Reuters Citation Laureate),2016年获得莱布尼茨奖(Gottfried Wilhelm Leibniz-Prize)。
1、突破传统金属催化局限,利用光氧化还原催化生成烯烃自由基阳离子,结合手性IDPi抗衡阴离子实现离子对导向的对映选择性控制,为烯烃环丙烷化提供绿色替代方案。
2、对苯乙烯、脂肪二烯、多烯烃等底物均表现高区域选择性(>20:1)和对映选择性(最高99:1 er),解决了多烯烃底物的区域选择性难题。
3、发现对映选择性依赖光波长,黄色光(长波长)可抑制重氮化合物直接光解的路径,增强催化路径占比,为光催化机制研究提供新探针。
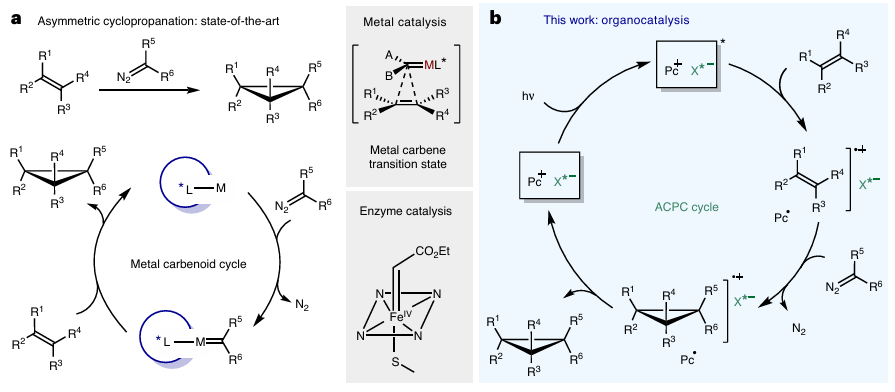
图1 烯烃和重氮化合物之间的不对称环丙烷化
图1对比了传统催化方法与本研究的反应机制。图1a展示了基于金属催化剂(如铜、铑配合物)或工程化酶(如细胞色素P450)的经典路径,首先由金属或酶活化重氮化合物生成亲电中间体,随后与烯烃发生[2+1]环加成形成环丙烷。该过程依赖金属中心的配位作用,虽能实现高对映选择性,但存在多烯烃底物区域选择性差、金属残留等问题。图1b为本研究提出的不对称抗衡阴离子导向光氧化还原催化(ACPC)机制,光激发阳离子通过单电子转移氧化烯烃,生成烯烃自由基阳离子,其与手性咪唑二磷酰亚胺阴离子形成离子对,引导重氮烷烃选择性插入碳碳双键,伴随氮气释放生成环丙烷自由基阳离子中间体,最后通过催化剂自由基的单电子转移再生PC⁺,得到目标产物。该机制通过手性离子对控制立体化学,避免金属参与,且适用于电子中性烯烃。
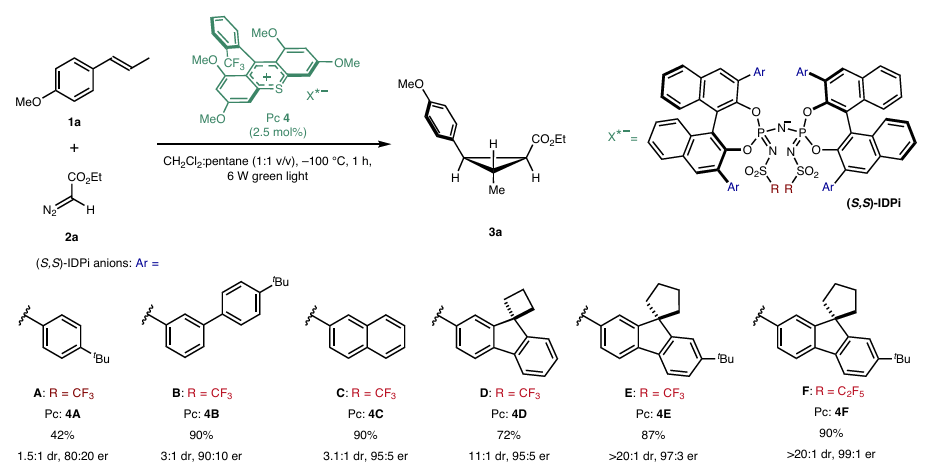
图2 反应过程
图2展示了手性IDPi阴离子的结构优化及反应性能评价。研究以反式丙烯基茴香醚(1a)与重氮乙酸乙酯(2a)为底物,在-100 °C下通过绿色光照射筛选不同IDPi衍生物。IDPi-A实现中等产率和80:20 er,但非对映选择性较差。IDPi-B通过扩大空间位阻提升产率至90%,er改善至90:10;进一步引入IDPi-E后,dr显著提升至20:1,表明大位阻基团可增强非对映选择性控制;最终优化的IDPi-F(核心为全氟乙基磺酰胺)将er提高至99:1,且在0.5 mol%催化剂负载下仍实现完全转化。
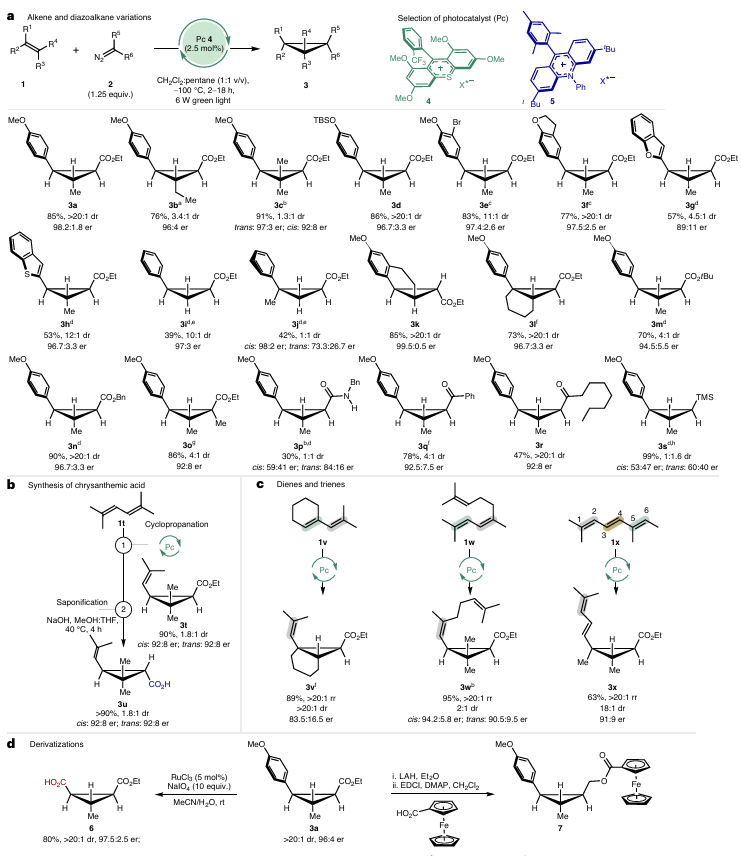
图3 环丙烷化的底物范围
图3展示了该催化体系的底物普适性与实用性。图3a显示,多种烯烃底物均能高效反应,含乙基、三甲基硅氧基的苯乙烯能够实现97:3 er以上。杂环烯烃表现优异的对映选择性,电子中性的苯乙烯需更高氧化电位的催化剂在红光下反应,er达95:5。内烯烃(3k-l)和不同重氮酯(3m-o)均适用,甚至含酰胺、酮基的重氮烷烃(3p-r)也能生成目标产物。图3b展示了二烯烃衍生物的应用,2,5-二甲基己二烯(1t)在无需过量底物的情况下,选择性生成单环丙烷化产物3t(dr≈3:1,er 90:10),水解后得到菊酸(3u),且保留立体选择性。图3c进一步展示多烯烃的区域选择性,不对称共轭二烯实现>20:1区域选择性。图3d通过衍生化反应验证产物实用性。
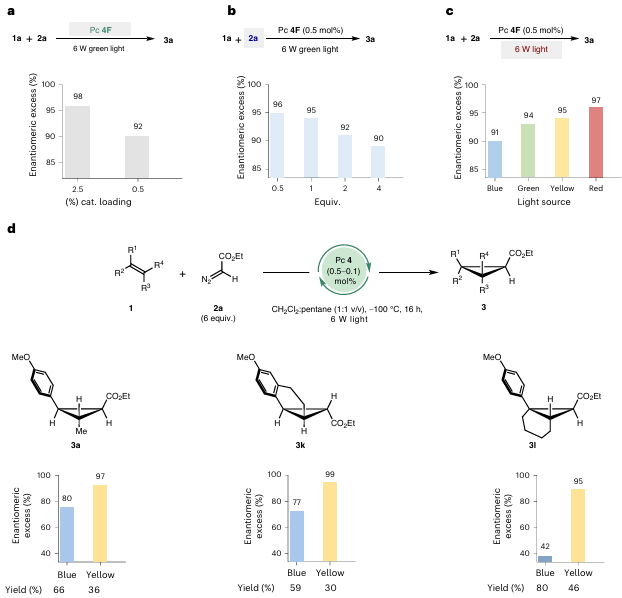
图4 光波长对反应的影响
图4揭示了光波长与对映选择性的关联性。图4a显示,降低催化剂负载至0.5 mol%时,er从99:1略微下降至96:4,表明低催化剂浓度下非催化反应占比增加。图4b中,过量重氮酯导致er降低。图4c-d通过不同光源实验发现,长波长光源(黄色/红色)显著提升er,反应在黄色光下er从80%提升至97%,内烯烃底物(1l)的er从41.8%提升至95.4%。结合光谱分析,催化剂4F的吸收峰(λmax=472 nm)与重氮酯2a的吸收峰(λmax=397 nm)部分重叠,短波长光(如蓝光)可能激发重氮酯直接光解。
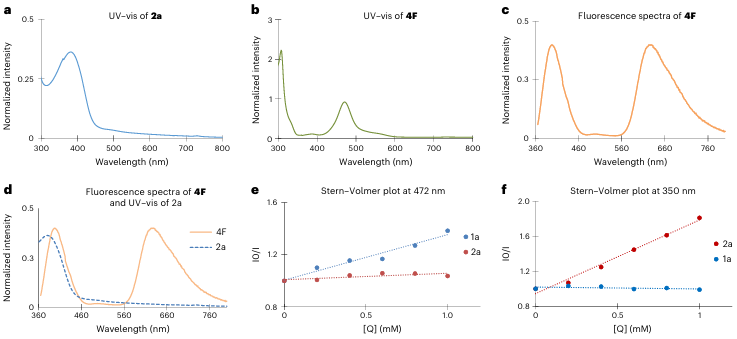
图5 机理分析
图5展示了本文中关键的光谱分析实验,用于揭示反应机制。图5a和图5b分别展示了重氮酯(2a)和光催化剂(4F)的紫外-可见吸收光谱,其中重氮酯在397 nm处有最大吸收峰,而光催化剂4F在472 nm处有最大吸收峰。图5c展示了光催化剂4F的荧光发射光谱,当在350 nm激发时,除了常见的627 nm发射峰外,还检测到一个395 nm的额外发射峰,这表明光催化剂在不同波长激发下具有不同的反应模式。图5d将重氮酯的吸收光谱与光催化剂的发射光谱叠加,发现两者的吸收带和发射带存在显著重叠,表明重氮酯可能通过光催化剂的低波长激发态参与反应。图5e和图5f展示了光催化剂4F在不同激发波长下的荧光猝灭实验结果。当在472 nm激发时,产物1a显著猝灭了光催化剂的荧光,而重氮酯(2a)几乎无影响;而在350 nm激发时,重氮酯则显著猝灭了荧光。
本文通过不对称反离子导向光氧化还原有机催化(ACPC)实现了烯烃的高对映选择性环丙烷化反应,解决了多烯烃底物的区域选择性问题,并展示了广泛的底物适用性。这一方法有望在天然产物和药物分子的合成中发挥重要作用,尤其是在需要高对映选择性和区域选择性的复杂分子构建中。此外,波长依赖的对映选择性现象为光催化反应机制的研究提供了新的视角,可能启发更多基于光氧化还原催化的新型反应设计。
Organocatalytic regio- and stereoselective cyclopropanation of olefins. Nature Catalysis, https://doi.org/10.1038/s41929-025-01340-7.