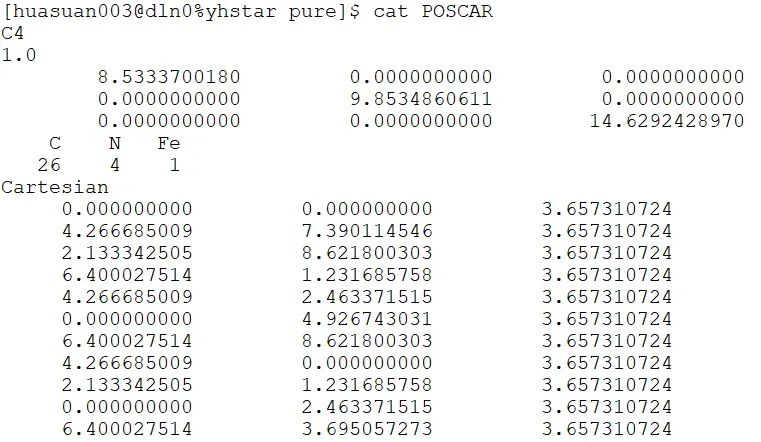
以VASP软件为例,首先通过晶体结构数据库下载材料的cif文件,或者在此基础上构建催化剂吸附H模型,并生成POSCAR文件,并根据元素顺序构建POTCAR文件,然后编写结构优化的INCAR和KPOINTS文件。准备好这四个文件后即可开始结构优化计算。
以VASP软件为例,首先通过晶体结构数据库下载材料的cif文件,或者在此基础上构建催化剂模型,并生成POSCAR文件,并根据元素顺序构建POTCAR文件,然后编写结构优化的INCAR和KPOINTS文件。准备好这四个文件后即可开始结构优化计算。
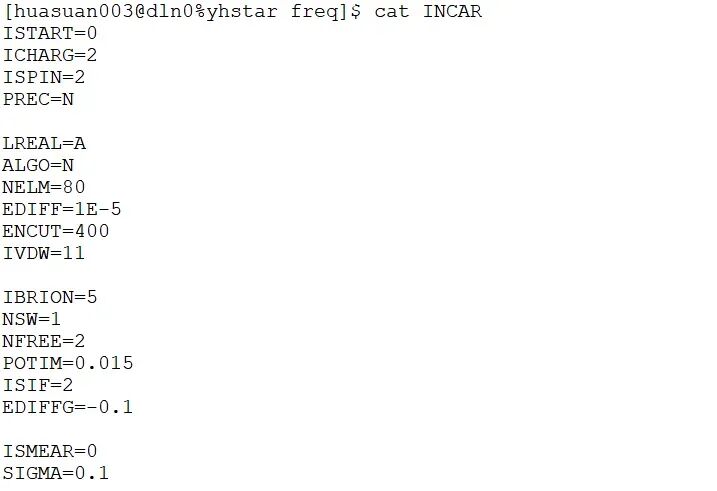
ICHARG=2 #从原子电荷密度产生体系初始电荷密度
EDIFF=1E-4 #电子波函数能量收敛标准1E-4 eV
IBRION=2 #共轭梯度法优化晶体结构和原子坐标
NSW=200 #晶体结构和原子坐标优化步数最大200步
EDIFFG=-0.1 #原子残余力小于0.1 eV/A
ISMEAR=0 #费米能级附近电子占据数为高斯分布
Automatic generation #注释行
完成结构优化计算后,保持KPOINTS,POTCAR文件不变,将CONTCAR文件复制成POSCAR文件,并对结构优化的INCAR文件作如下修改,从而获得体系的总能E
完成结构优化计算后,保持KPOINTS,POTCAR文件不变,将CONTCAR文件复制成POSCAR文件,并对结构优化的INCAR文件作如下修改:
在POSCAR中固定催化剂原子,也就是在原子坐标后加上FFF,放开吸附的原子或分子,也就是在原子坐标后加上TTT,并加上选择动力学标签。
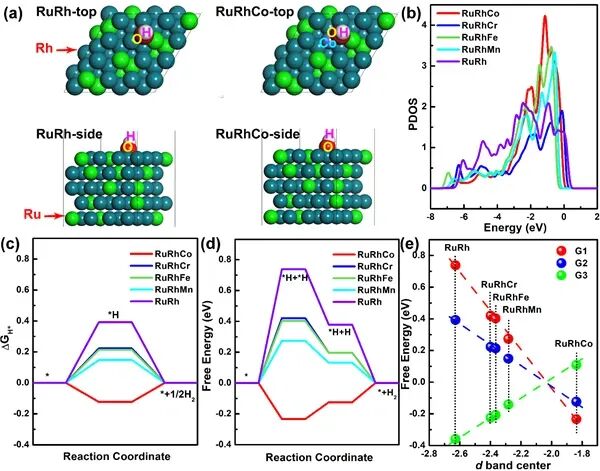
完成振动计算后,下载原子振动频率计算的OUTCAR文件,用脚本获得ZPE和TS,并得到吉布斯自由能G=E+ZPE-TS
?500+博士团队护航,累计助力5️⃣0️⃣0️⃣0️⃣0️⃣➕篇科研成果,计算数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等国际顶刊。 ???
声明:如需转载请注明出处(华算科技旗下资讯学习网站-学术资讯),并附有原文链接,谢谢!